PFP Validation for public v5.0.0
Overview
Since the release of Matlantis in July 2021, PFP (Preferred Potential) [1], the core NNP (Neural Network Potential ) technology, has been updated about once every six months. and as of February 2024, the latest version, v5.0.0, is available. Here we present the results of the verification of the latest version, v5.0.0.
Results
Benchmarks with Matbench Discovery
Matbench Discovery [2] is a benchmark designed to explore new stable inorganic crystals. Using structures generated by elemental substitution of a structure from the Materials Project [3] (WBM Dataset [4]) as input, Matbench Discovery performs structural optimization and computes formation energies, etc., and compares them with DFT (Density Functional Theory) results.
It is important to note that while Matbench Discovery covers some actinides from Ac to Pu, PFP does not support these elements. Therefore, structures containing these elements are excluded from the results below. In addition, there are differences between the DFT conditions in Matbench Discovery and the DFT conditions in the PFP dataset. Corrections have been made to absorb these differences. Please refer to the [Details] chapter for details on the correction.
Matbench Discovery shows the results of training the Materials Project [3] dataset on multiple architectures as a leaderboard. Matbench Discovery focuses on the phase diagram at zero temperature and zero pressure. In a phase diagram, the energy difference from the convex hull, which consists of a group of crystal structures that are thermodynamically stable, is called energy-above-hull, and is used to determine the stability of a crystal structure. The ROC (Receiver Operating Characteristic) curve, which shows the relationship between TPR (true positive ratio) and FPR (false positive ratio), is drawn at different thresholds for determining stability. The ROC curve is shown below.
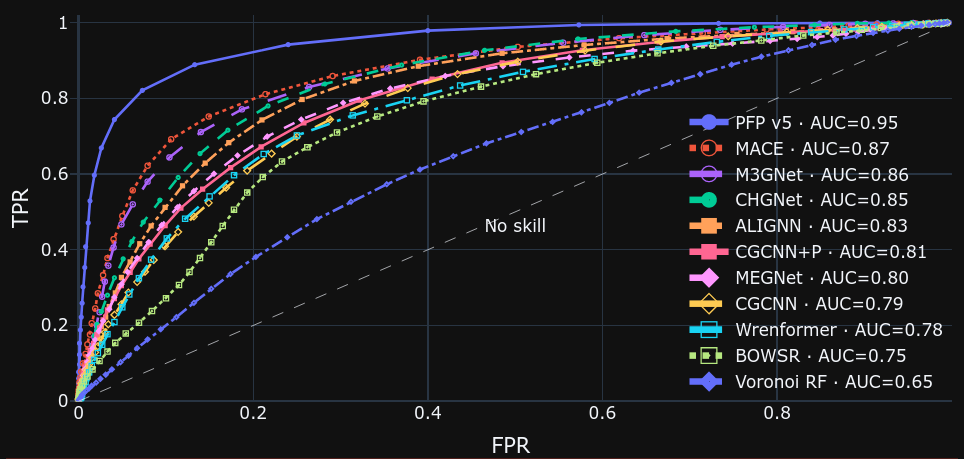
The ROC curves show that the further away we go left from the x=y straight line, the better the predictions, indicating that PFP v5.0.0 outperforms the other models. This is probably due to the fact that PFP v5.0.0 is trained on a more diverse and larger dataset compared to the Materials Project dataset.
The element-specific MAEs of energy above hull for a given elemental structure are compared between PFP v5.0.0 and MACE, the top performing model on the leaderboard.
| PFP v5.0.0 | MACE |
 | 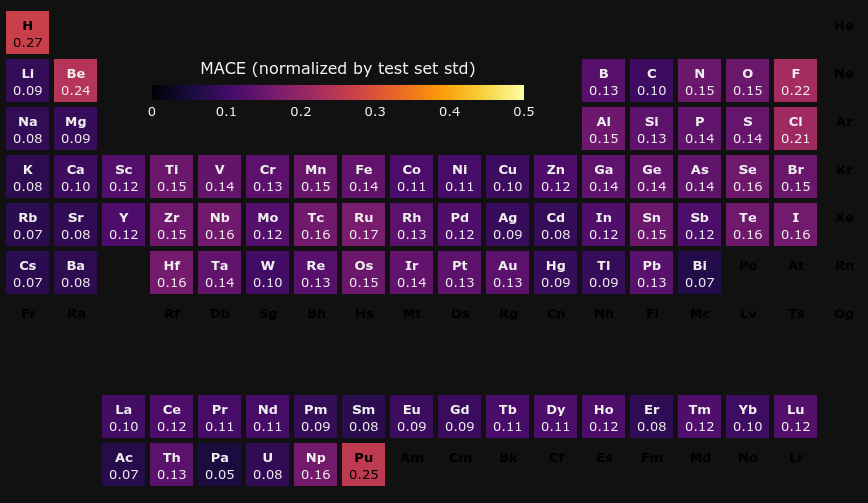 |
In PFP v5.0.0, there are no elements with extremely large MAEs, and it can be said that PFP has general-purpose potential for various elements. Yb has a larger MAE than MACE, but we believe this is because the pseudo potential of Yb is chosen differently between the PFP dataset and the Materials Project.
Other performance indicators shown in Matbench Discovery are listed below. See [2] for definitions of the indicators. In all cases, PFP v5.0.0 scores high and excels in the task of searching for new stable inorganic crystals. The values for the other models refer to the values of the leaderboard on the official web page on February 14, 2024.
| F1 ↑ | DAF ↑ | Prec ↑ | Acc ↑ | TPR ↑ | TNR ↑ | MAE ↓ | RMSE ↓ | R2 ↑ | |
| PFP v5.0.0 | 0.76 | 5.26 | 0.75 | 0.92 | 0.77 | 0.95 | 0.03 | 0.07 | 0.84 |
| PFP v5.0.0 (72elem) | 0.77 | 8.16 | 0.76 | 0.93 | 0.79 | 0.96 | 0.03 | 0.07 | 0.85 |
| MACE | 0.674 | 3.1378 | 0.584 | 0.885 | 0.80 | 0.896 | 0.06 | 0.10 | 0.6770 |
| CHGNet | 0.61 | 3.3609 | 0.512 | 0.854 | 0.764 | 0.876 | 0.06 | 0.10 | 0.69 |
| M3GNet | 0.57 | 2.8867 | 0.445 | 0.810 | 0.8077 | 0.81 | 0.07 | 0.121 | 0.5860 |
PFP v5.0.0 (72elem) is the score of PFP when narrowed down to only the supported 72 elements.
The indicators in the table are explained individually. The indicators on the right side of the table, MAE, RMSE, and R^2, are quantities associated with regression errors; the smaller the overall deviation, the better the evaluation.
PFP also shows good scores for F1, Prec, Acc, TPR, and TNR, which are based on the binary classification of whether the energy value of the crystal structure is positive or negative with respect to the convex hull. On the other hand, since most of the data points are located in the vicinity of the convex hull, they are considered to be relatively sensitive to the DFT calculation conditions of the source dataset.
Since DAF tends to show higher values when structures that have not been computed are included, we omit an explanation here.
Reproducibility Verification of Crystal Volumes
In PFP v4.0.0, volumes of organic crystals were overestimated compared to DFT calculations. However, the situation has been greatly improved in PFP v5.0.0.
The figure shows a comparison of volumes calculated by DFT and PFP for structures obtained from the COD (Crystallography Open Database) [5]. It is known that dispersion forces have a significant effect on the volume of organic and complex crystals, so the D3 correction [6,7] of Germin et al was added. For inorganic crystals, no D3 correction was added.
| Relative volume | Relative error | |
| Organic crystals | 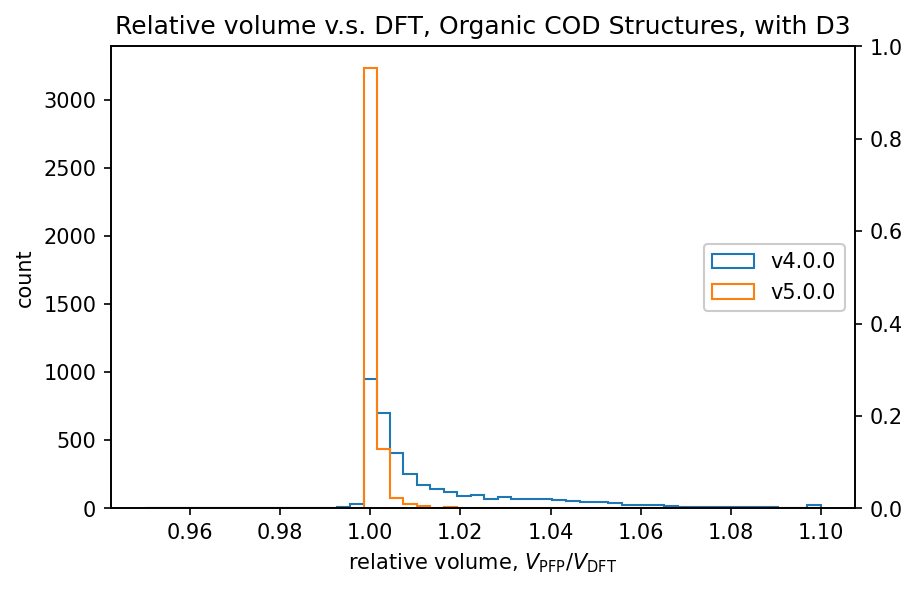 | 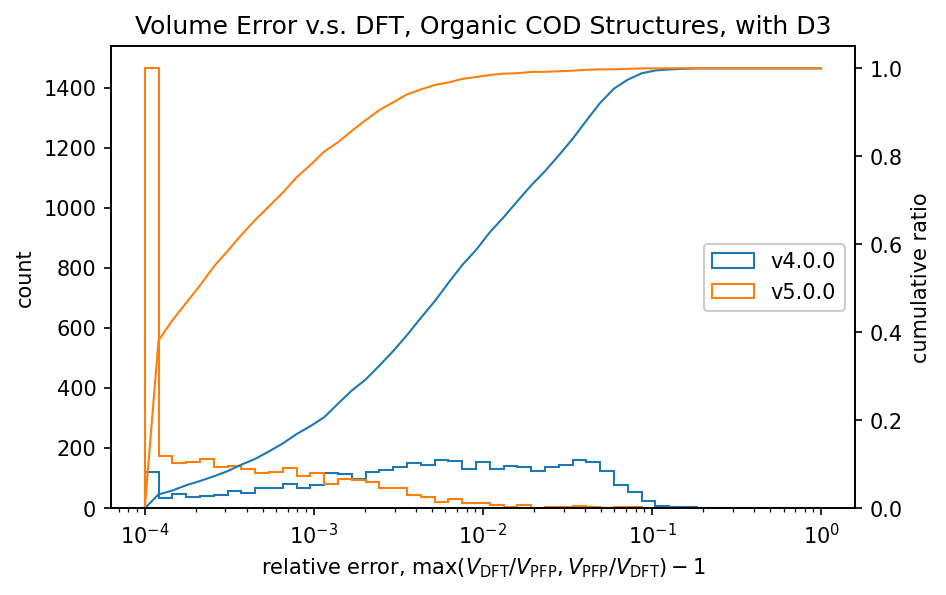 |
| Complex crystals | 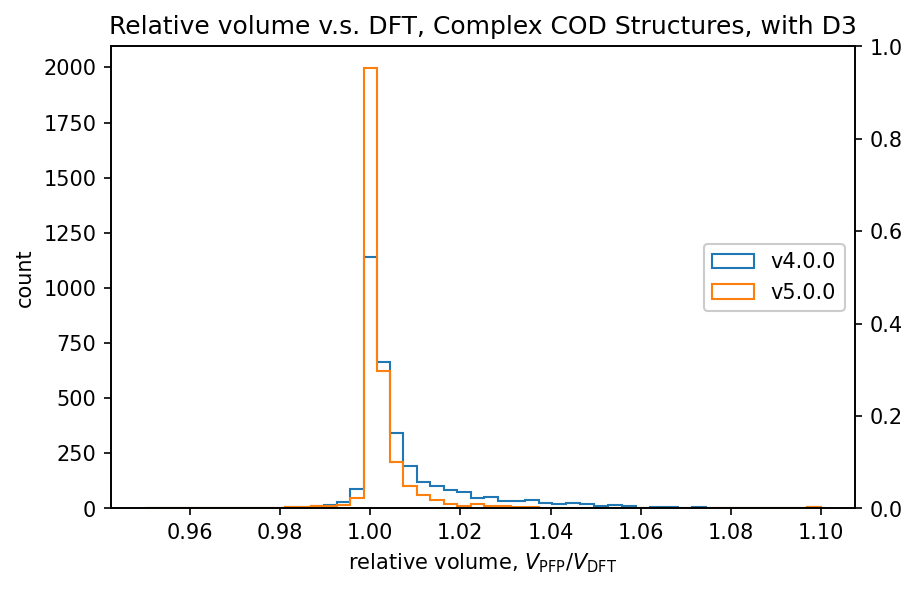 | 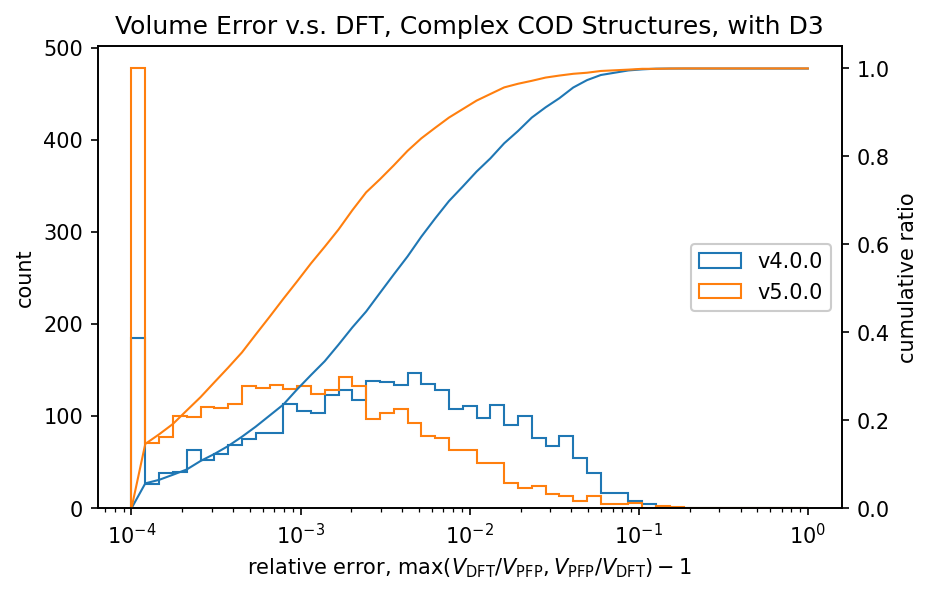 |
| Inorganic crystals | 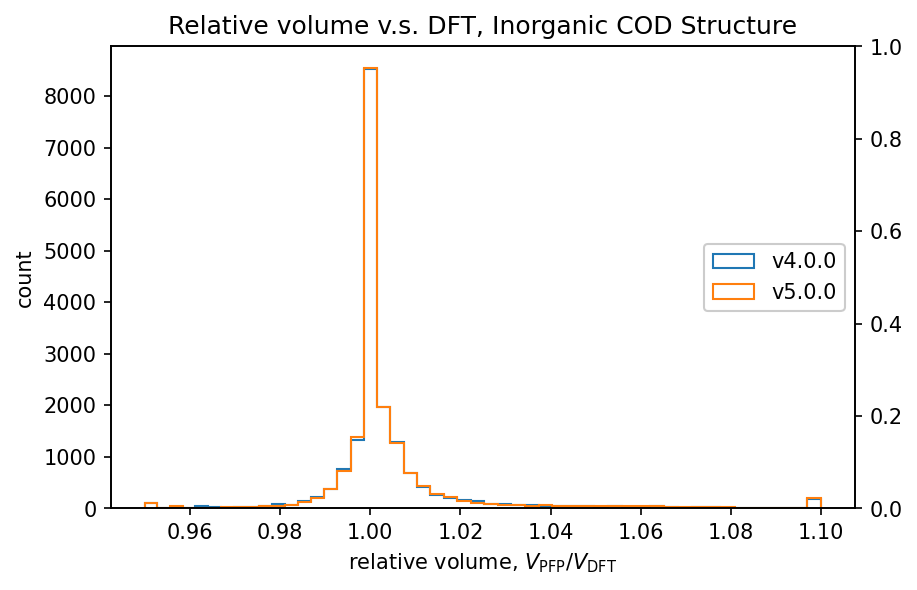 | 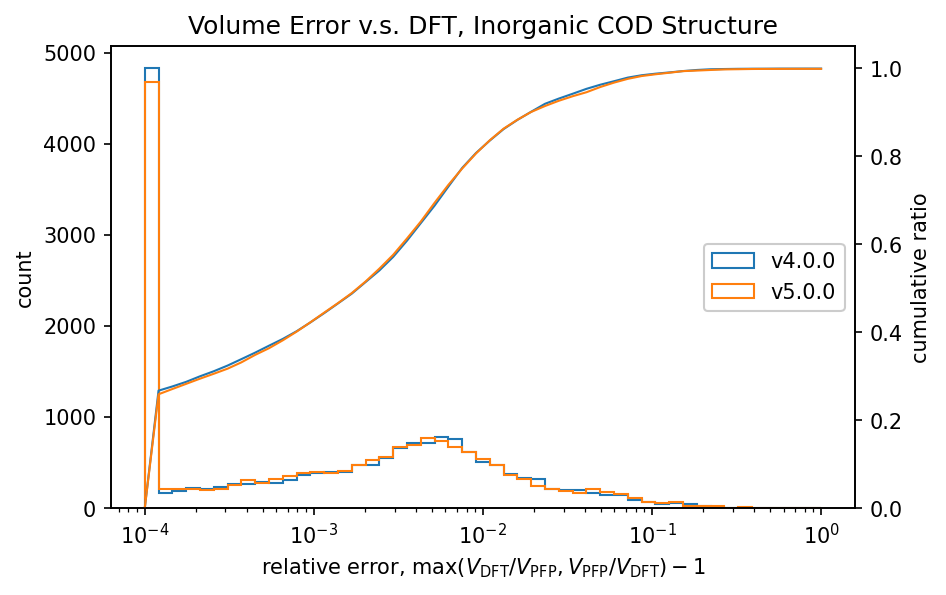 |
Histograms of relative volumes for organic crystals show that PFP v4.0.0 tended to overestimate volumes relative to DFT, but that this trend has been greatly improved in PFP v5.0.0. In addition, PFP v5.0.0 was able to determine the volume of 90% of the organic crystal structures with high accuracy, within a relative error of 0.23%. On the other hand, PFP v4.0.0 had a relative error of within 4.4%, which is a significant improvement in v5.0.0. For complex crystals, the volume reproducibility has also improved in PFP v5.0.0 compared to v4.0.0, although the improvement is not as great as for organic crystals. For inorganic crystals, the reproducibility is comparable.
In PFP v5.0.0, the reproducibility of potential energy surfaces for organic and complex crystals has been greatly improved by expanding the dataset. This is believed to have led to the improved reproducibility of volumes in organic and complex crystals. Although the aforementioned Matbench Discovery is mainly for inorganic crystals, these results indicate that PFP is a versatile potential that is also highly reproducible for organic and complex crystals.
Summary
Benchmark results from Matbench Discovery show that PFP v5.0.0 outperforms existing models in the task of searching for new stable crystals. This is likely due to the fact that it is trained on a more diverse and larger dataset.
PFP v5.0.0 has improved reproducibility of organic and complex crystals compared to PFP v4.0.0. We believe that this is due to the expansion of datasets and other factors, and the improvement in reproducibility of organic crystals is particularly noticeable. In addition, PFP is a versatile potential with high reproducibility for organic and complex crystals as well as inorganic crystals, which are the target of Matbench Discovery.
Details
This section provides details on the calculation methodology and additional information.
Benchmarking with Matbench Discovery
Matbench Discovery’s DFT conditions are based on those of the Materials Project, which are largely the same as the PFP dataset, but differ in smearing methods and other aspects. In order to absorb the differences between the DFT conditions, we use the Materials Project dataset ( 2023-02-07-mp-computed-structure- entries.json.gz ) created with the same DFT conditions as the WBM dataset of Matbench Discovery, and fitted the energy of each individual element to this dataset with least-squares fitting so that the formation energies match. This dataset is the training dataset in the Matbench Discovery benchmark, and this process does not leak any information from the test data.
Reproducibility Validation of Crystal Volumes
The DFT calculations for comparison were performed using VASP 5.4.4 or VASP 6.4.0. The differences between VASP 5.4.4 and VASP 6.4.0 were sufficiently small to be negligible. The PBE functional and the PAW (Projector Augmented Wave) method are used in the calculation. Please refer to the PFP paper [1] for more details. Note that PFP v2 or later offers a calculation mode without Hubbard correction, which is the subject of this verification. For this reason, Hubbard correction is not performed in the DFT calculations used for comparison.
Crystal structures were obtained from COD for crystal structures consisting of only 72 elements supported by PFP v3 or later, with a unit cell volume of 2200 ų or less. However, those with occupancies of less than 0.99 for each site, those with symmetry designation problems, etc. are excluded.
Organic, complex, and inorganic crystals were classified based on the following definitions
Organic crystals: Structures consisting only of H, C, N, O, P, S, F, Cl, Br, and I
Complex crystals: Structures that satisfy all of the following conditions
Containing at least two elements of H, C, N, O, P, S, F, Cl, Br, I
Containing at least one element other than H, C, N, O, P, S, F, Cl, Br, I
At least 80% of the atoms must be H, C, N, O, P, S, F, Cl, Br, or I
Inorganic crystals: Structures that are not classified as either organic or complex crystals.
For these crystal structures, DFT calculations were performed to optimize the structure, including cell parameters, so that the force on the atoms is less than 0.03 eV/Å, and a benchmark dataset was generated. As mentioned earlier, the organic and complex crystals were structurally optimized with D3 corrections.
The structure optimization in PFP was performed using the structure optimized by DFT as the initial structure, and the structure optimization including cell parameters was performed in PFP so that the force on the atoms is less than 0.03 eV/Å. It should be noted that there are many local minimums in organic crystals. In order to avoid comparing the volume of different local minimums as much as possible, the structure optimized by DFT is used as the initial structure.
Validation of energy and force for crystal structures
We believe that the reason for the improved volume reproducibility of organic and complex crystals is due to the improved reproducibility of potential energy surfaces for these structures. Below are the results of our evaluation of energy and force reproducibility.
For the above COD crystal structure, we performed structural optimization by DFT calculation including cell parameters so that the force on the atoms is less than 0.03 eV/Å. Then, we generated the structure with small displacement on the atomic positions. For details, please refer to “site position displacement” in Supporting Information, NOTE 10 of the PFP paper [1]. Note that the size of the displacement is smaller than in the paper. Note that, unlike the volume reproducibility test, no D3 correction is applied here, including structural optimization.
In the figure below, we compare the results of single-point calculations with DFT and PFP for an organic crystal structure with small displacements. v5.0.0 has about 1/3 of the MAE compared to v4.0.0, indicating a significant improvement in the reproducibility of the energy and force of the organic crystal.
| v5.0.0 | v4.0.0 | histogram | |
| Energy | 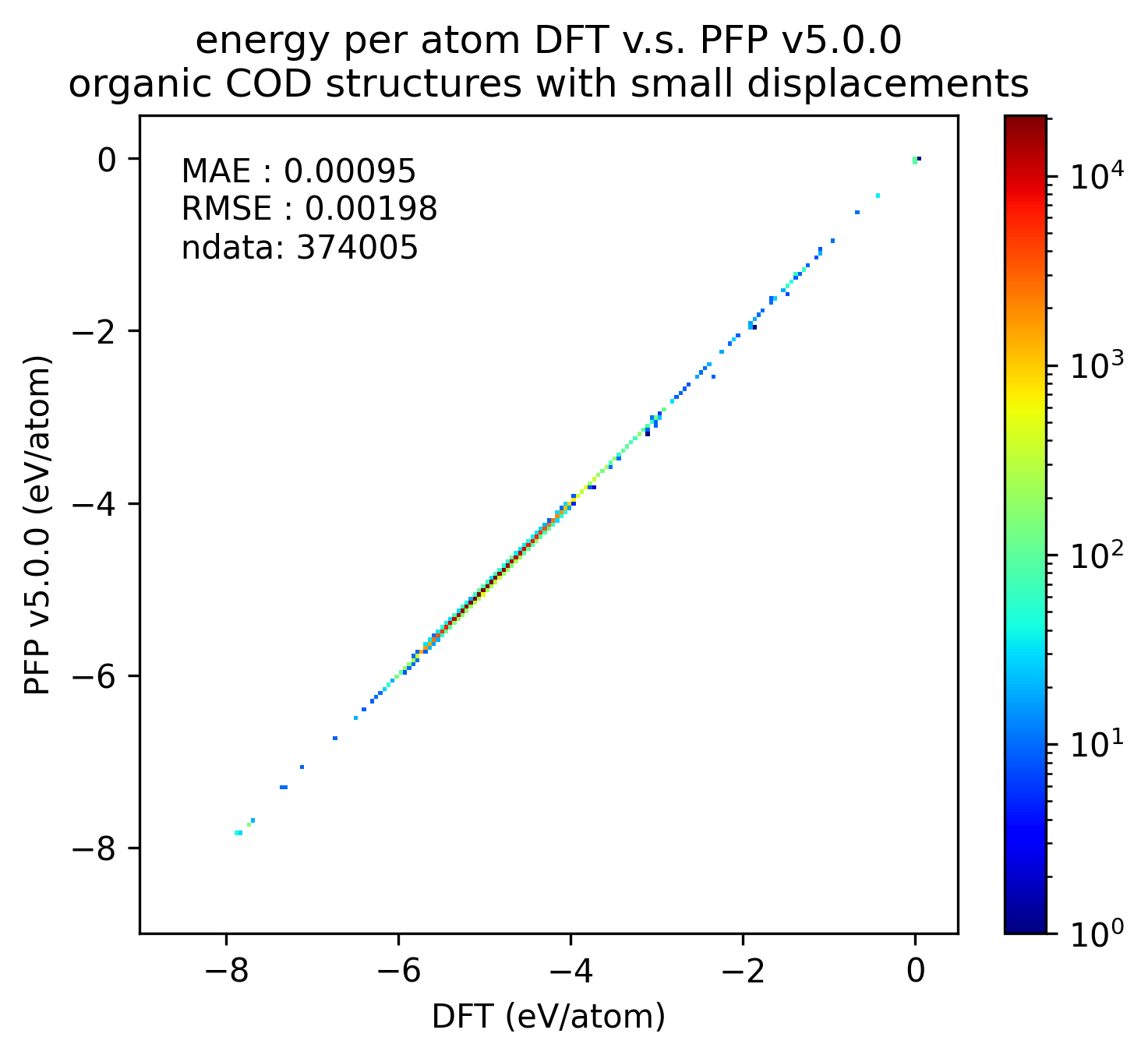 | 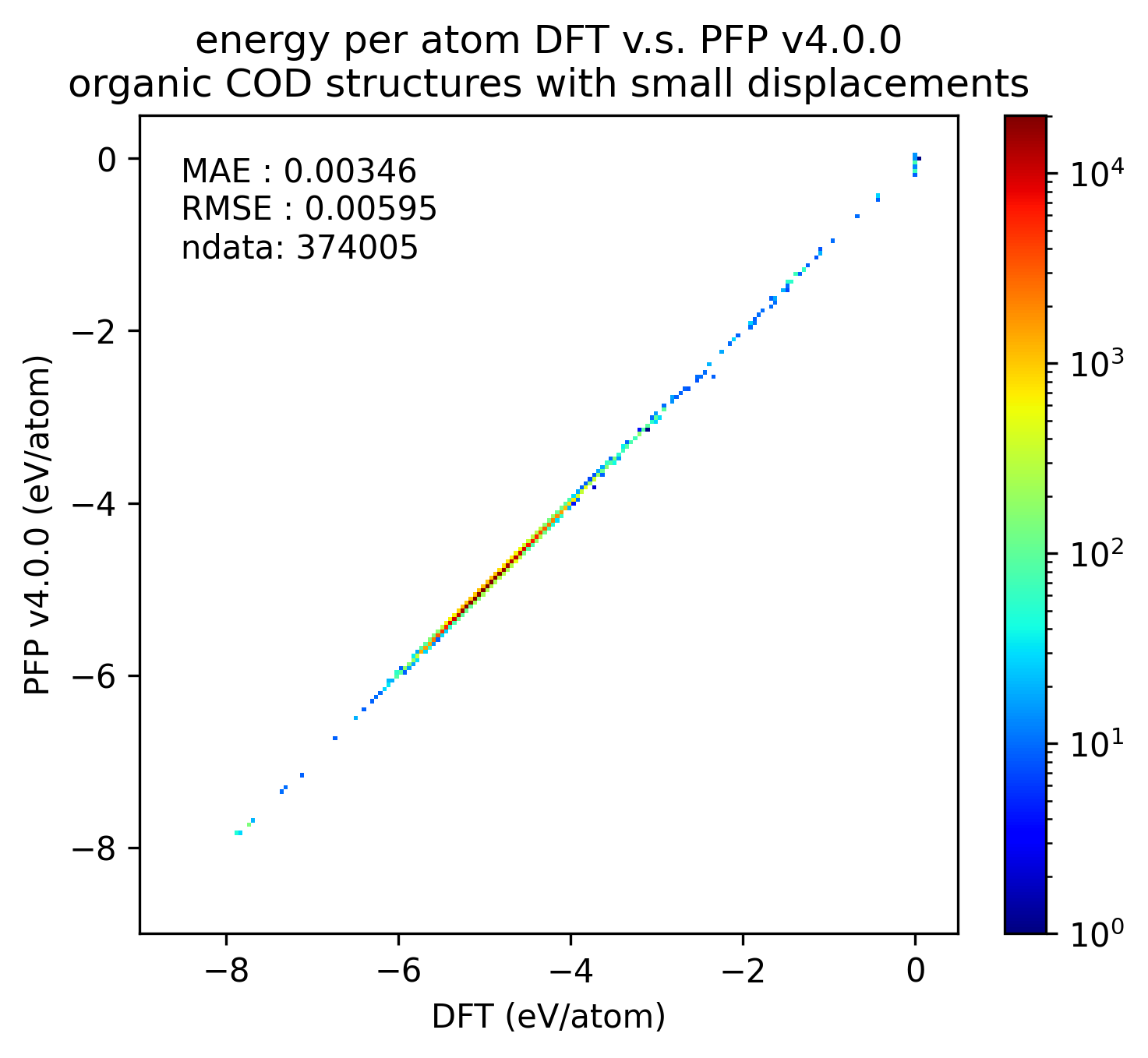 | 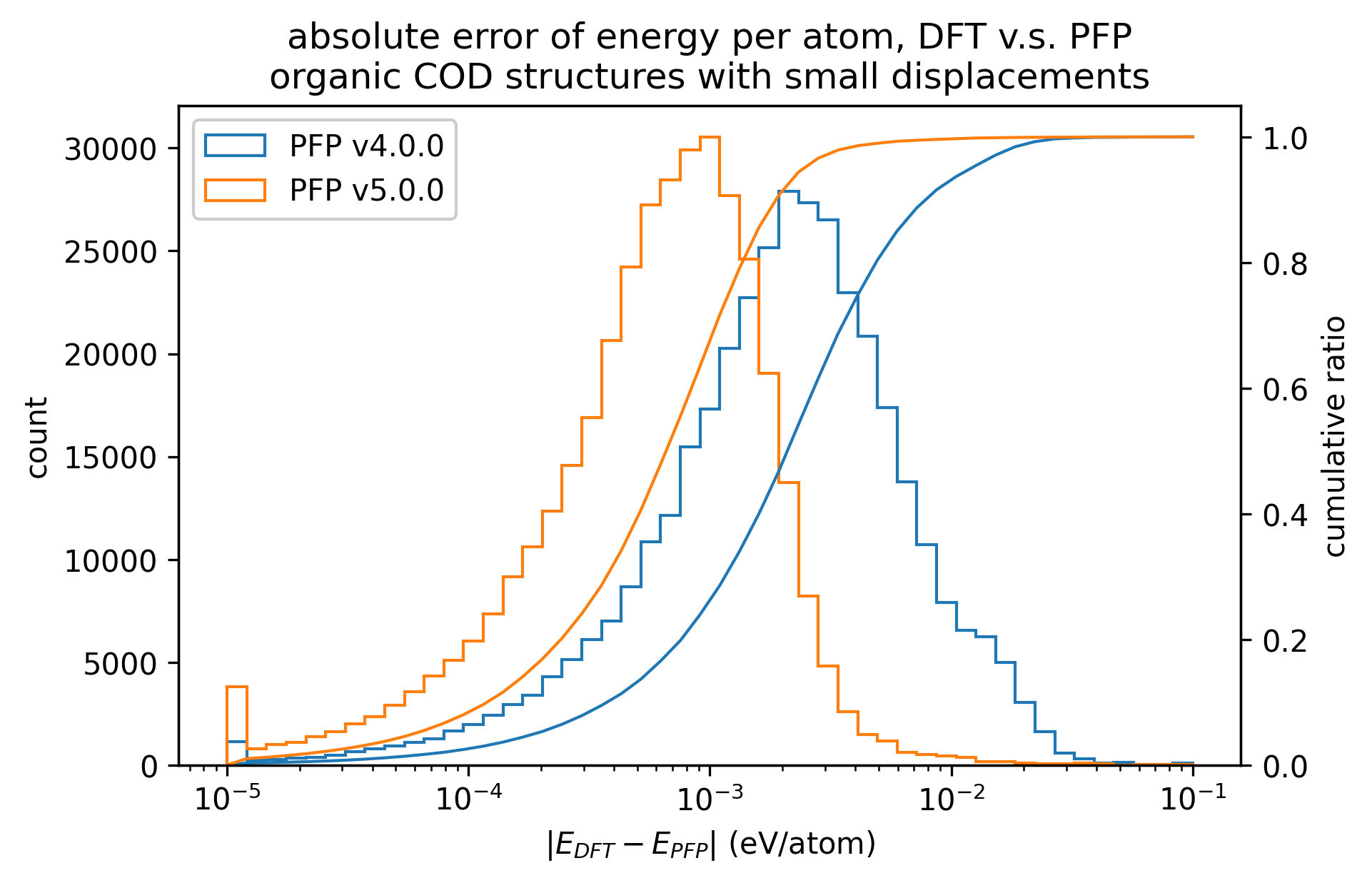 |
| Force | 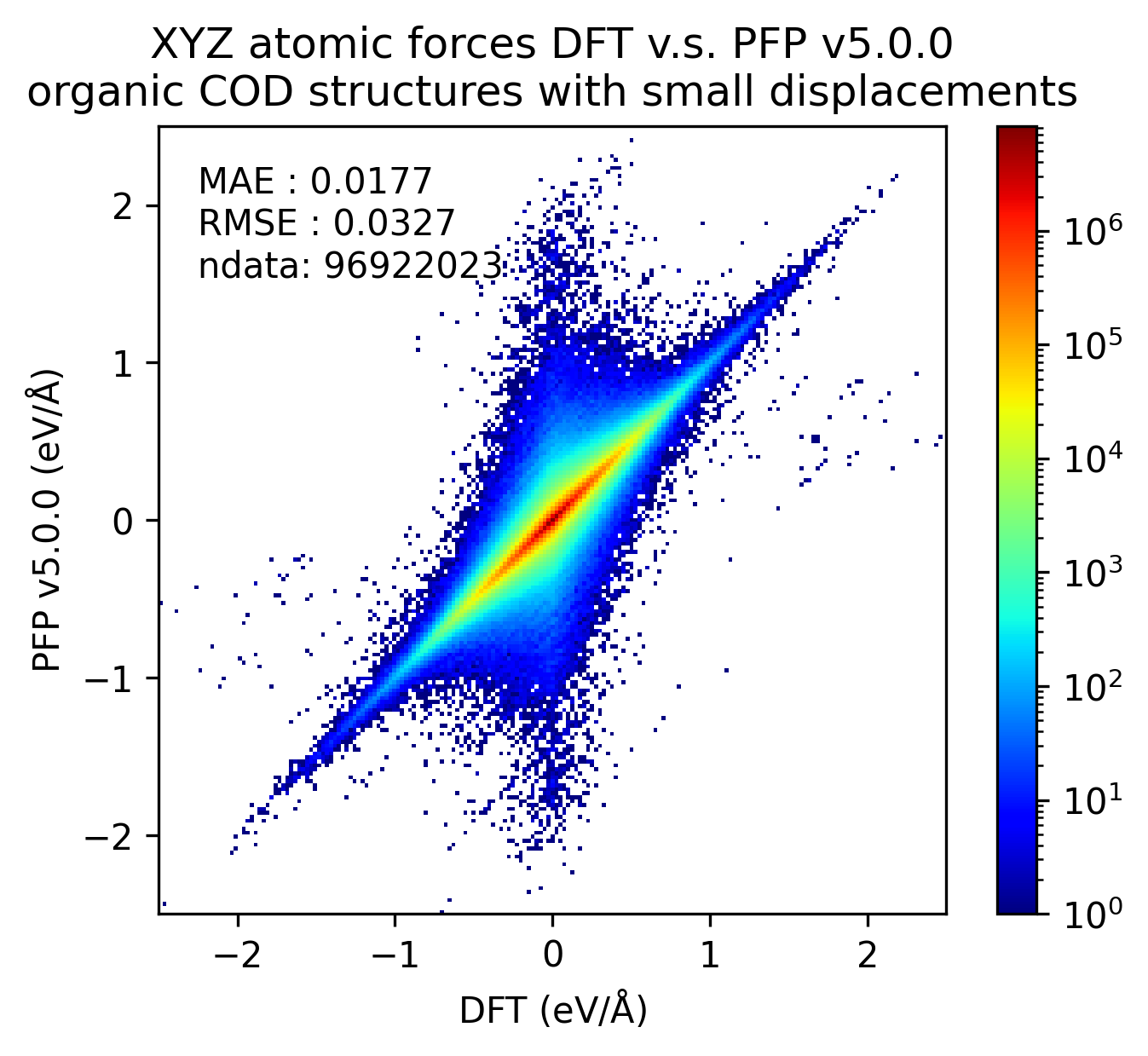 | 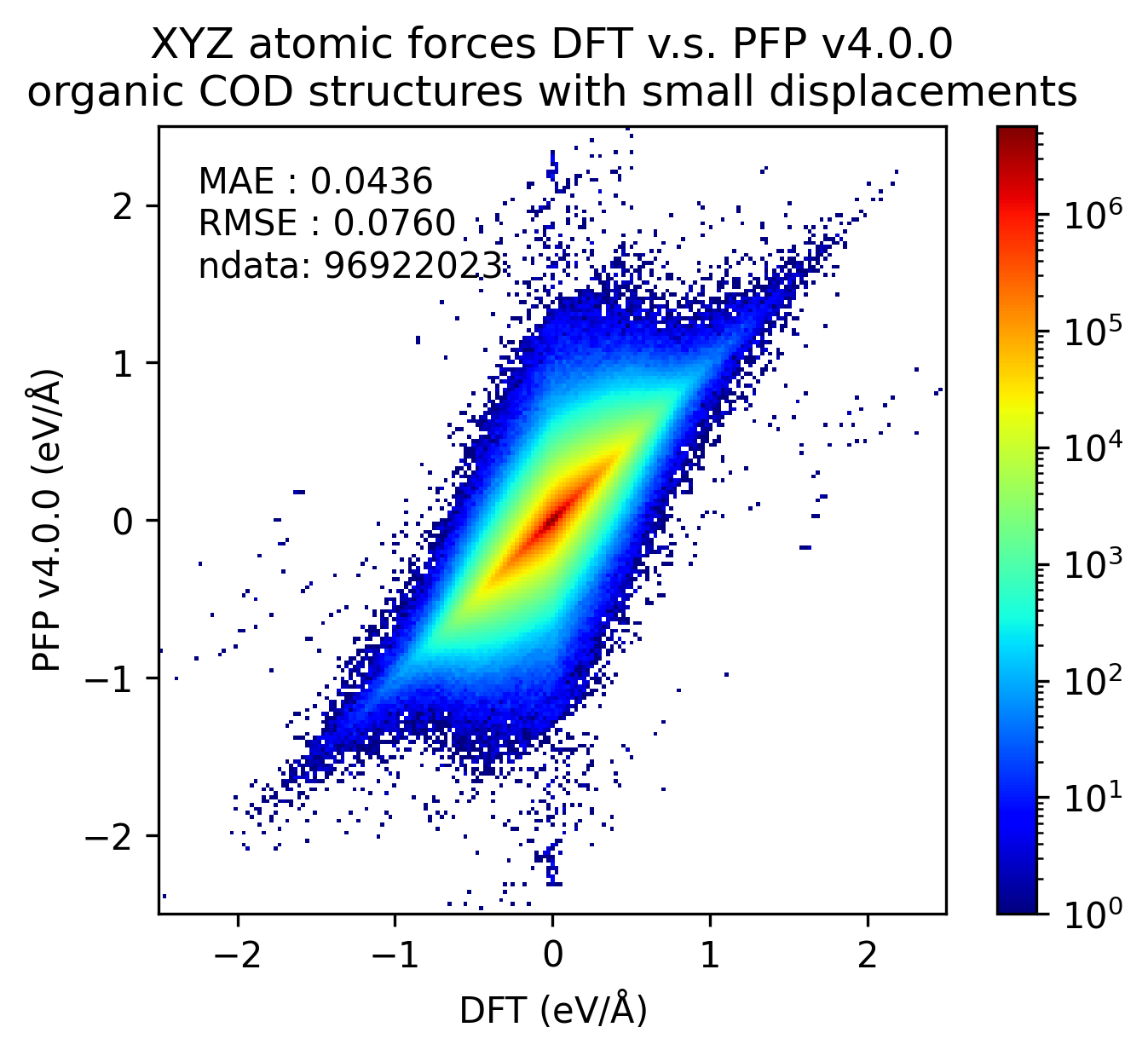 | 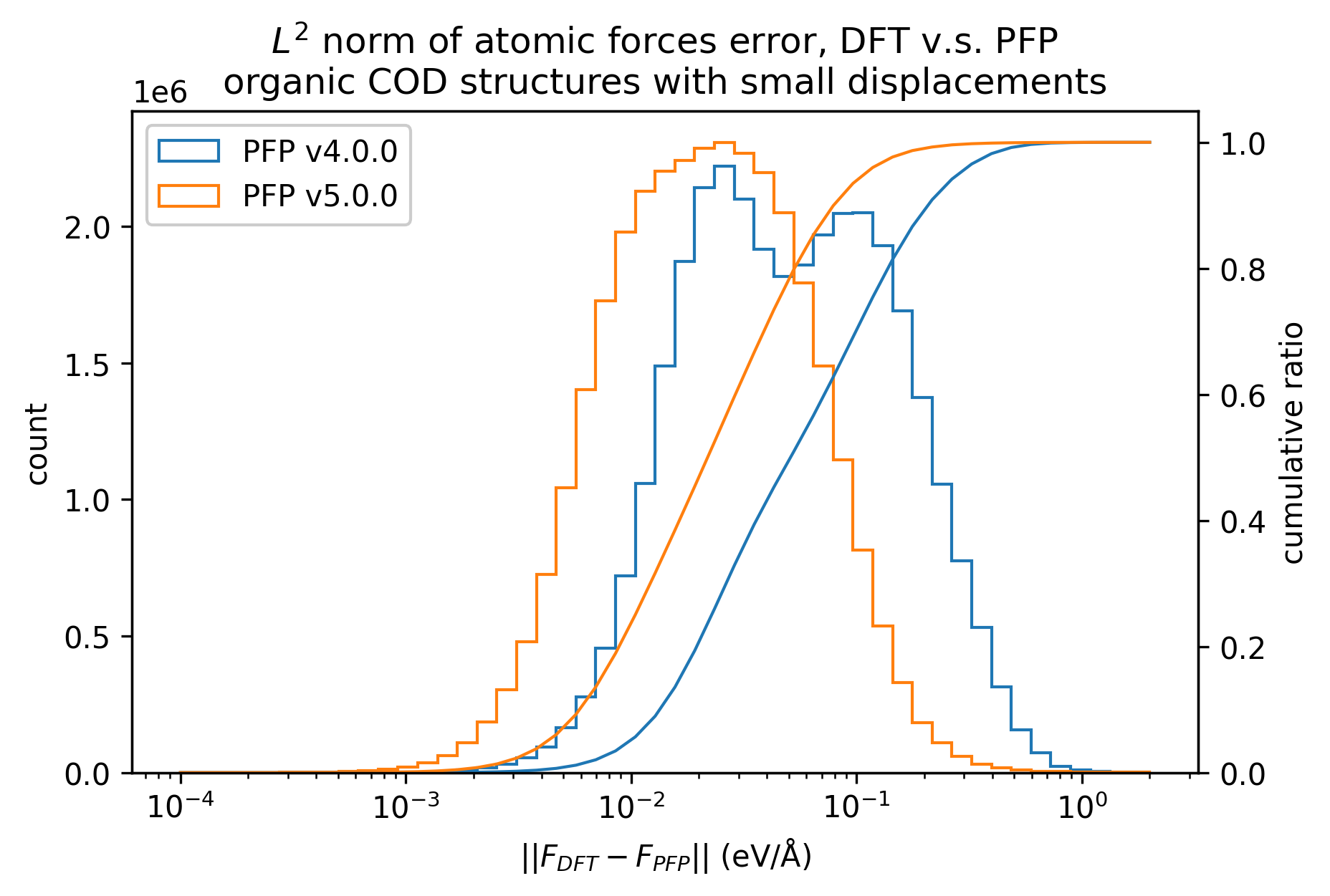 |
Similarly, the reproducibility of energy and force for complex crystals is shown.
Although not as pronounced as the results for organic crystals, we can see that the reproducibility of energy and force for complex crystals has improved in v5.0.0 compared to v4.0.0.
| v5.0.0 | v4.0.0 | histogram | |
| Energy | 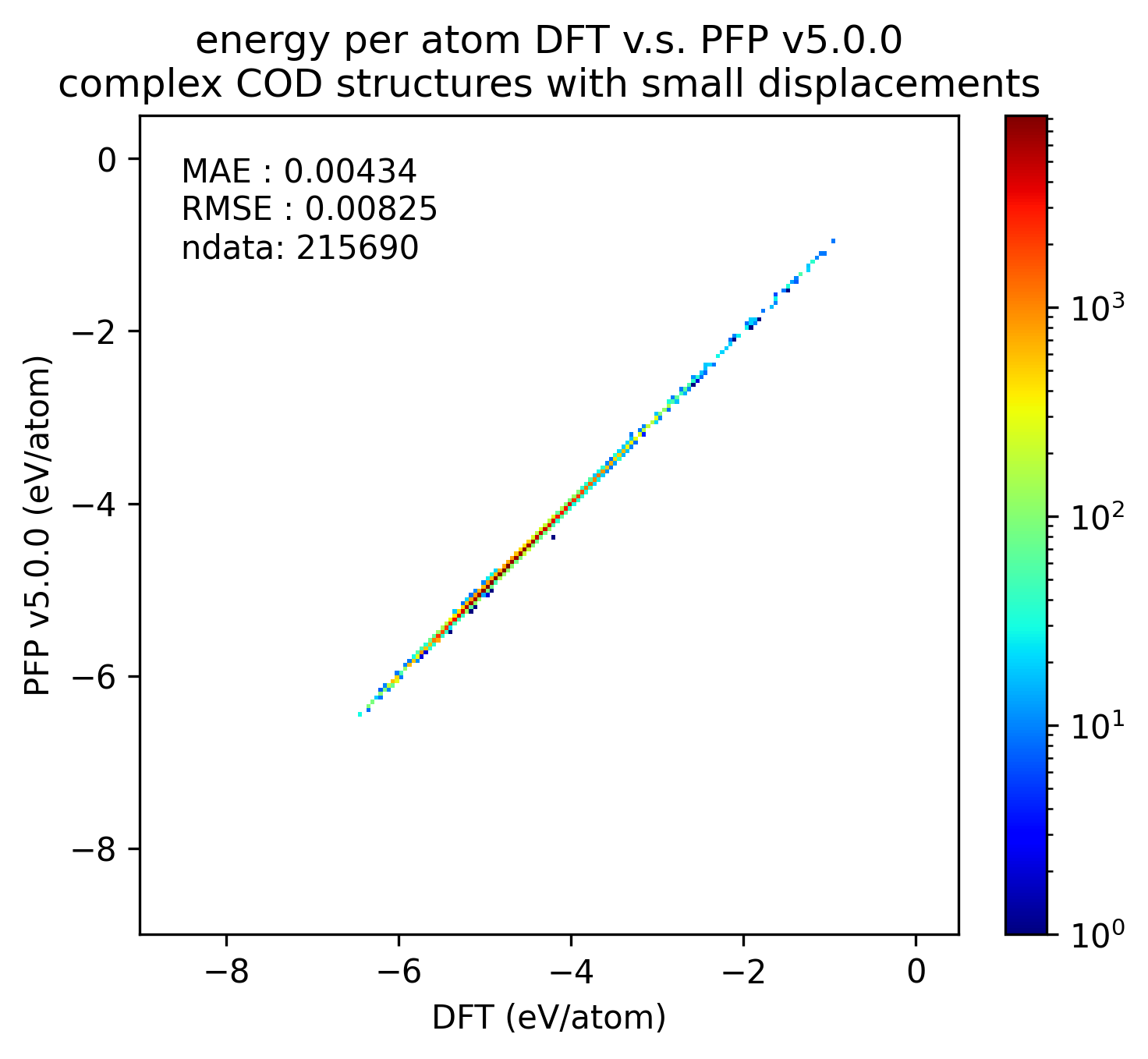 | 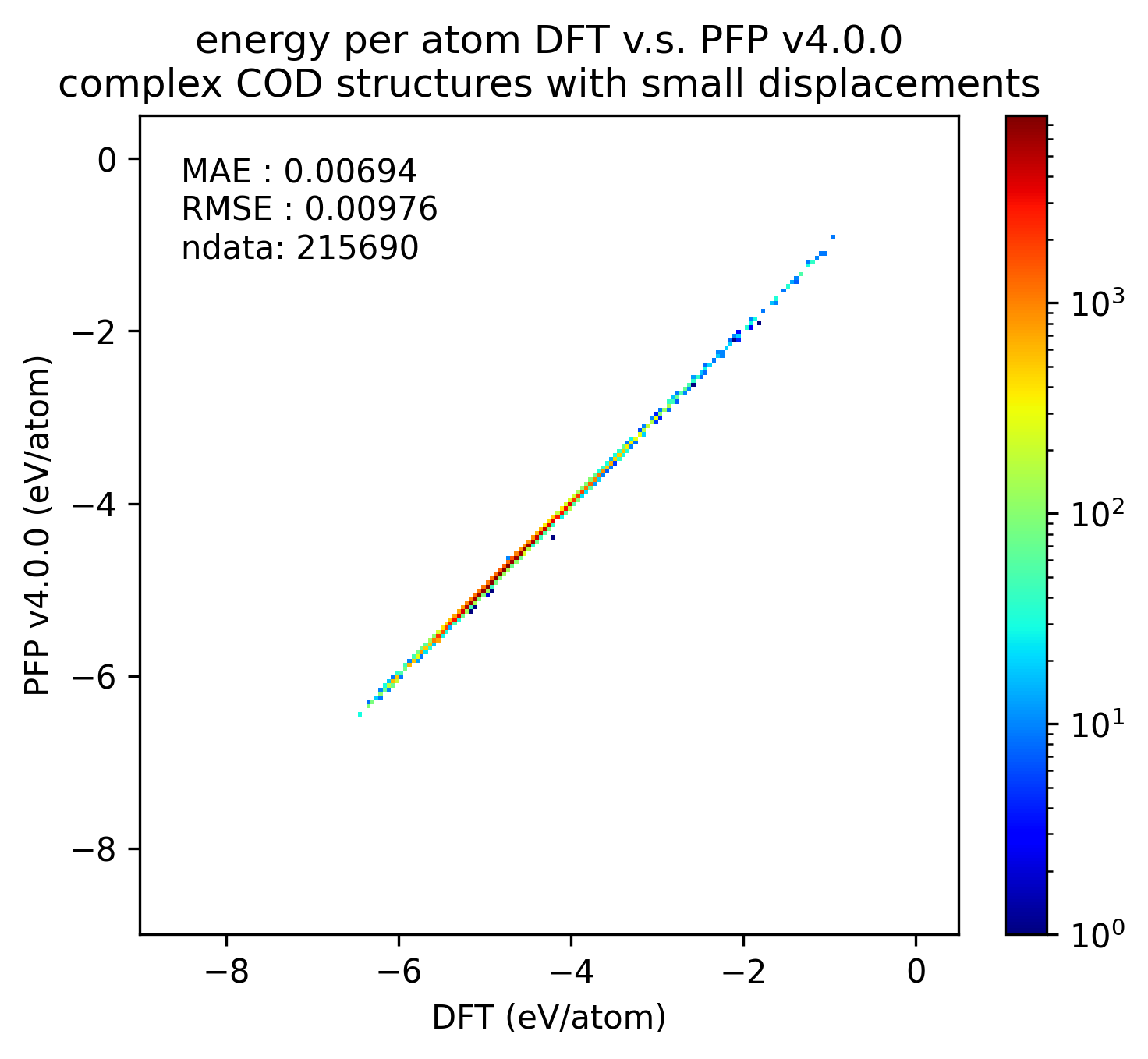 | 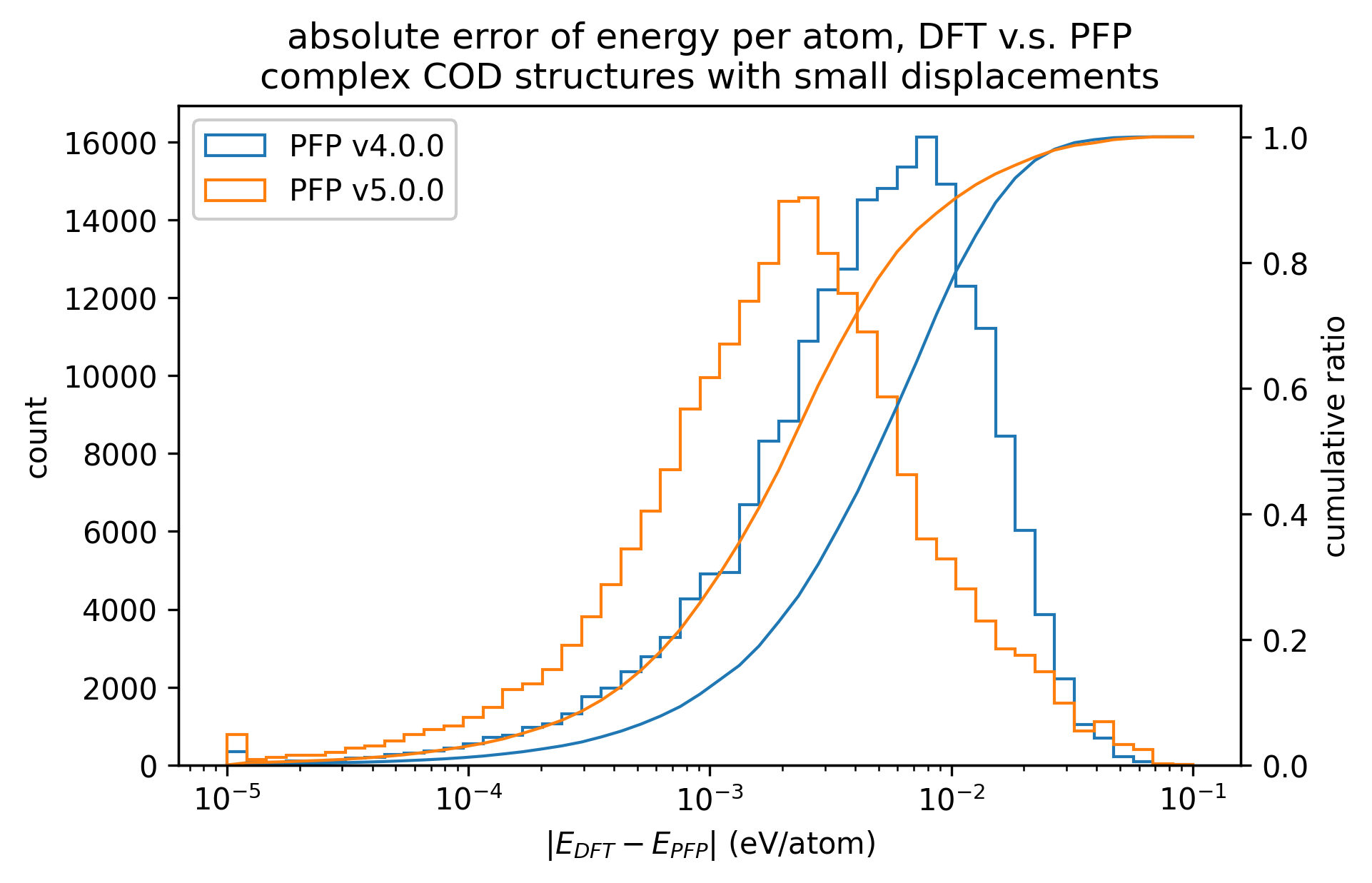 |
| Force | 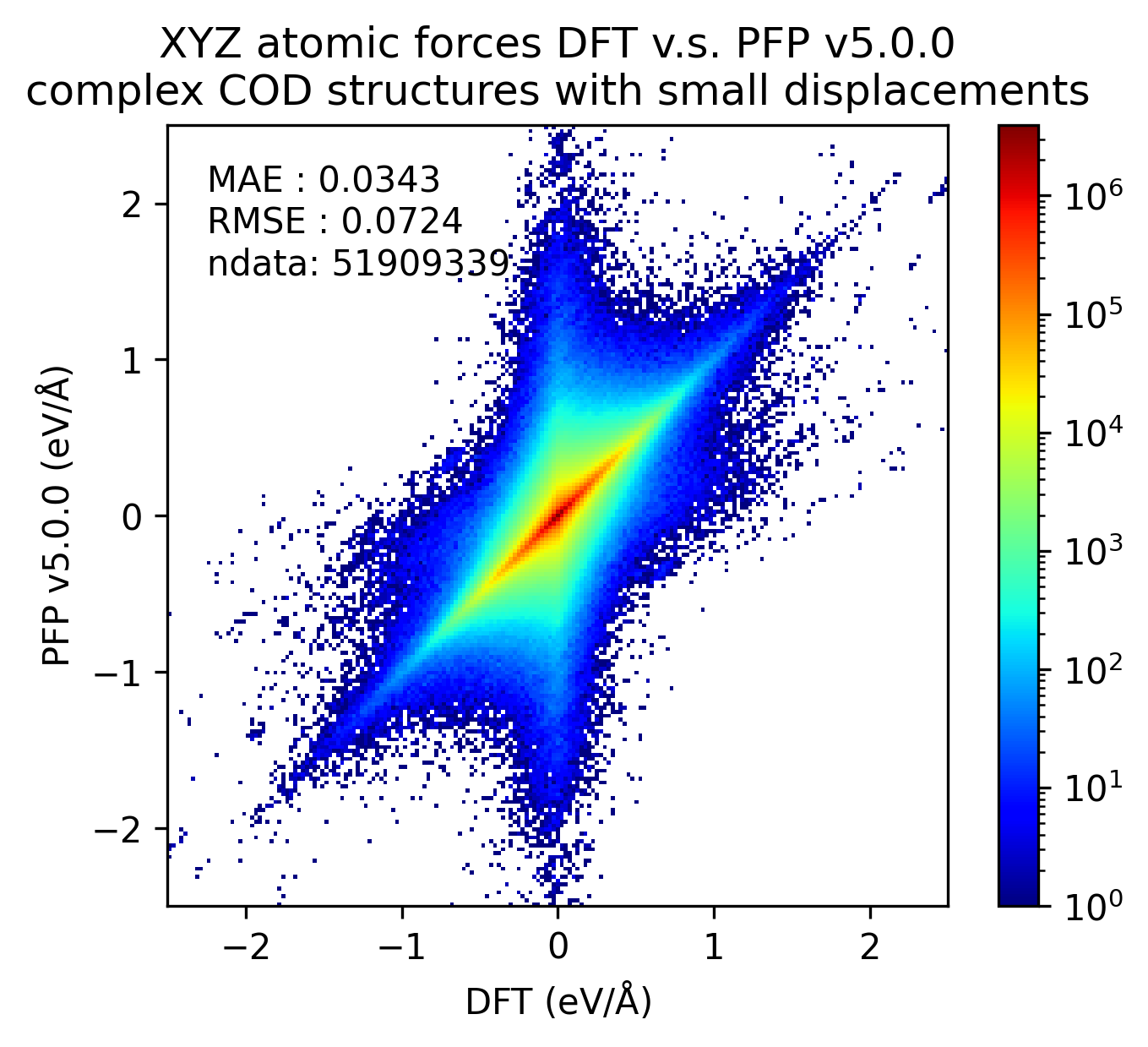 | 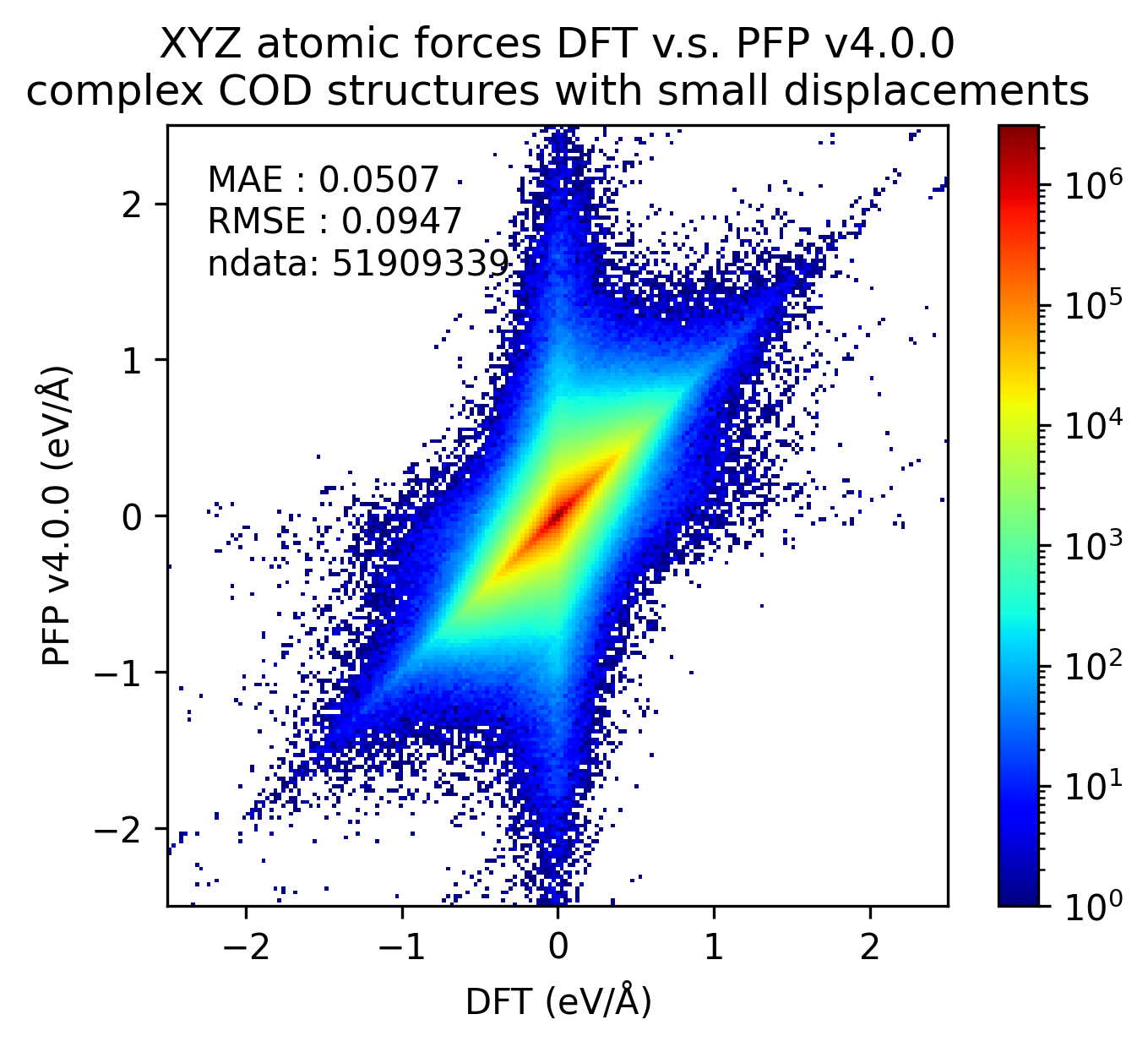 | 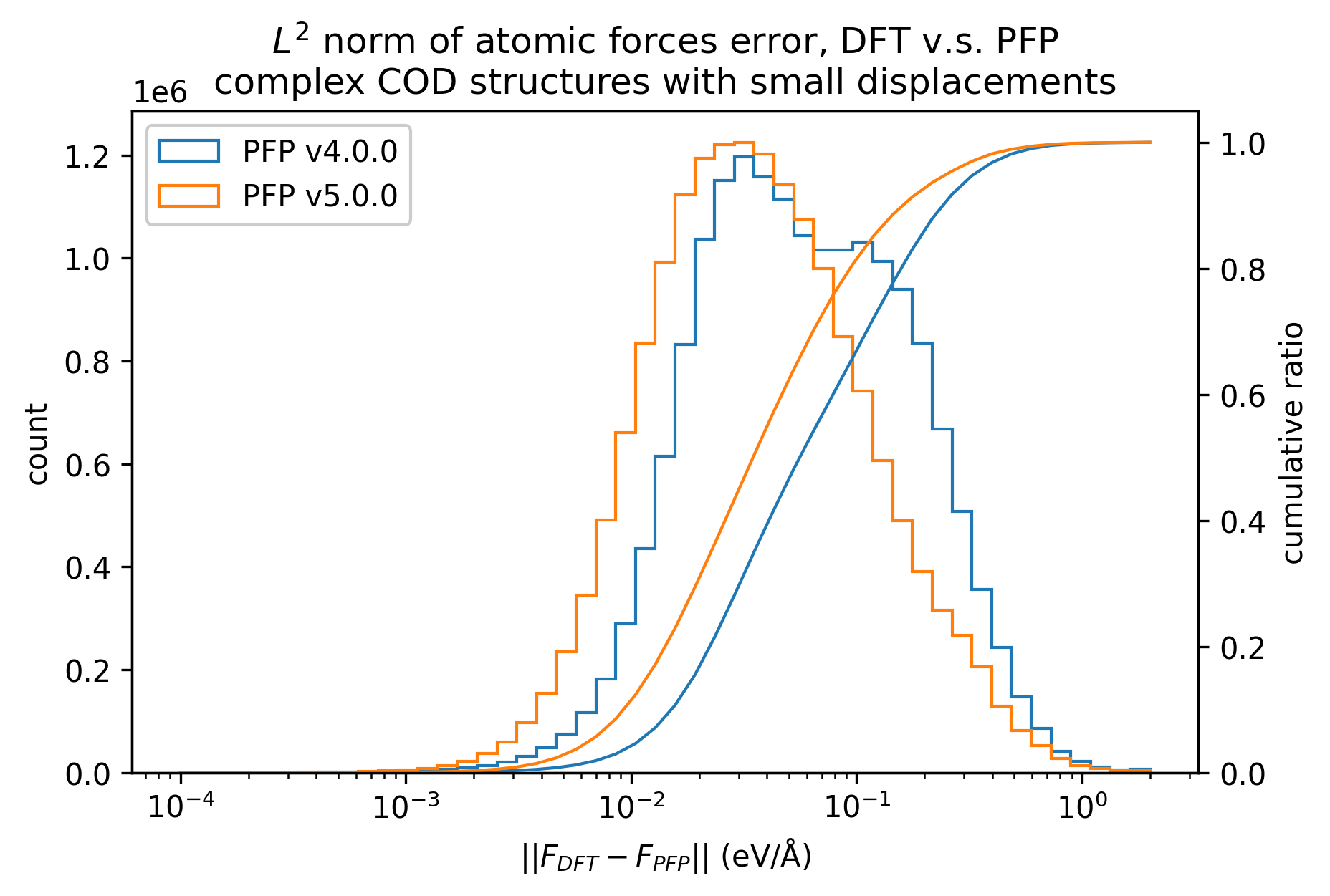 |
Reproducibility of inorganic crystals in terms of energy and force; MAE has slightly improved in PFP v5.0.0, but is still comparable.
| v5.0.0 | v4.0.0 | histogram | |
| Energy | 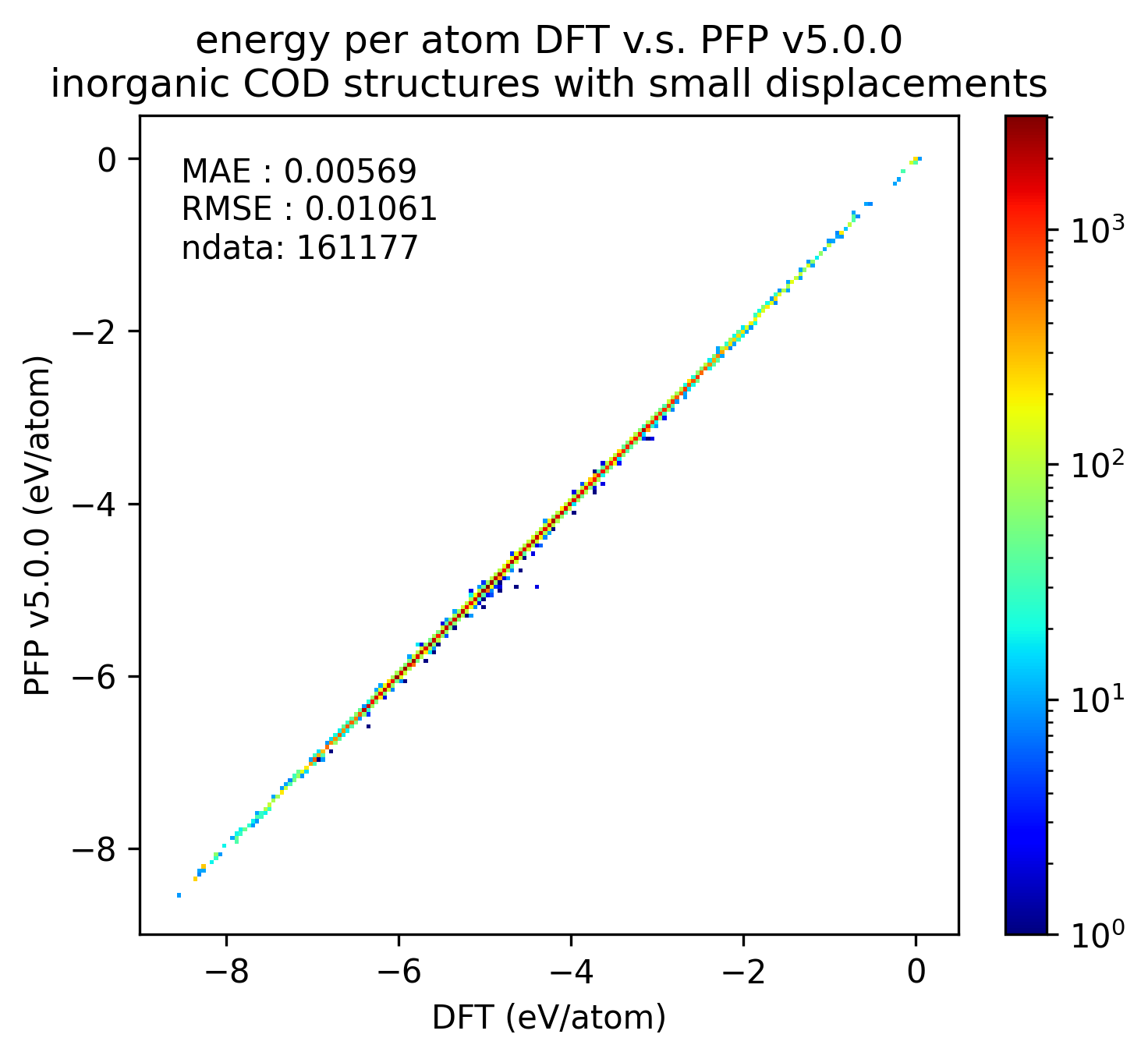 | 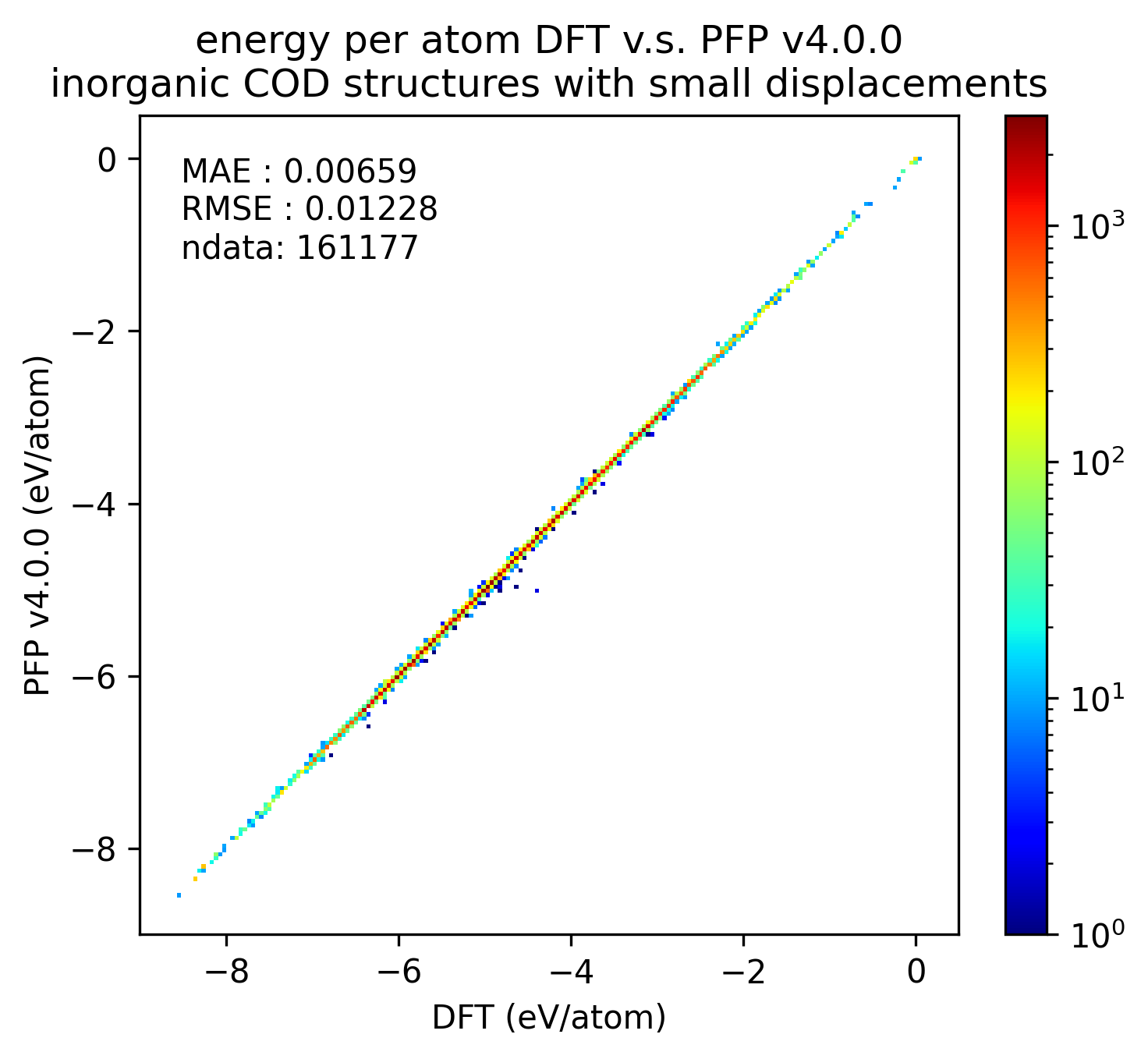 | 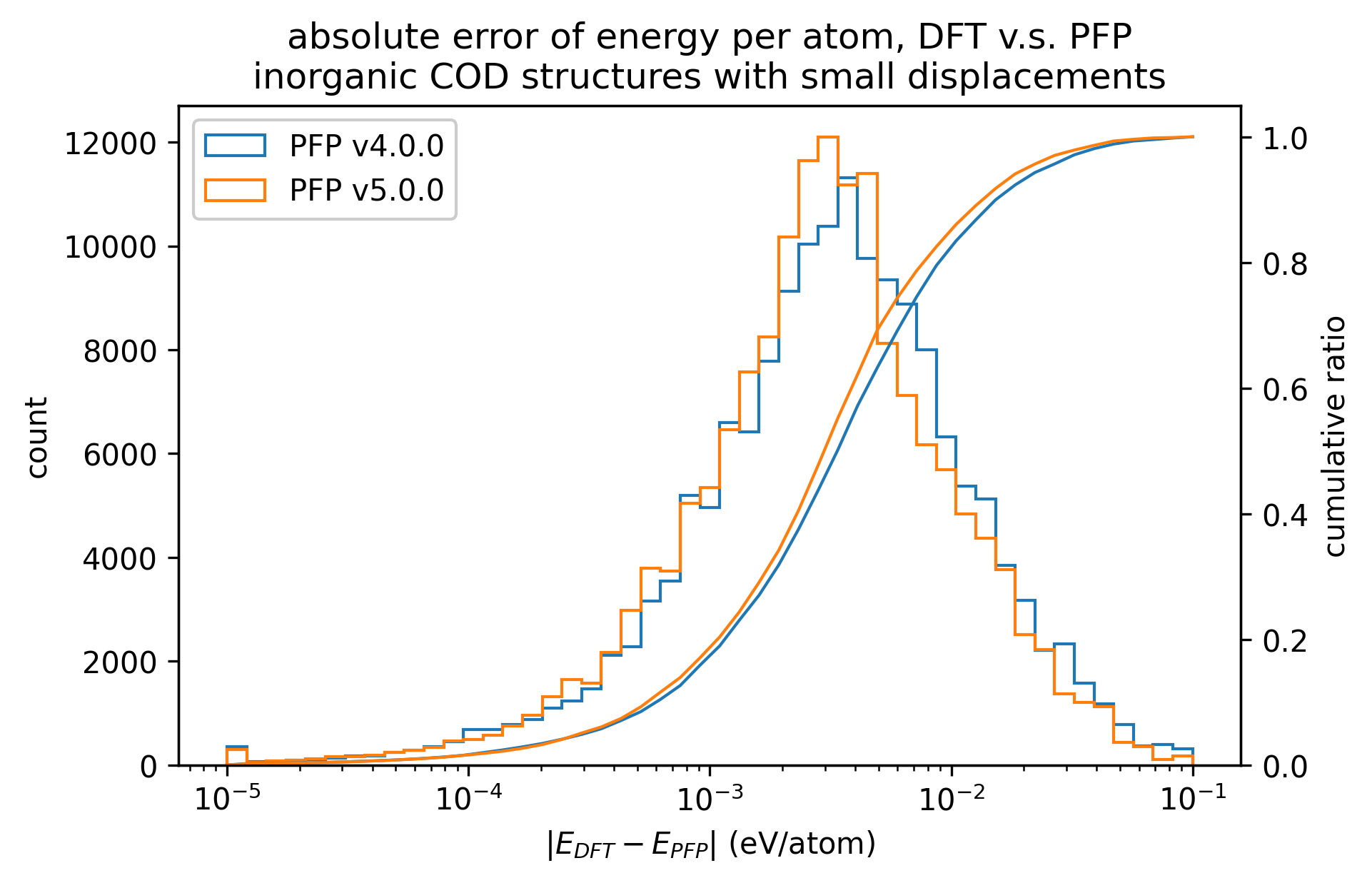 |
| Force | 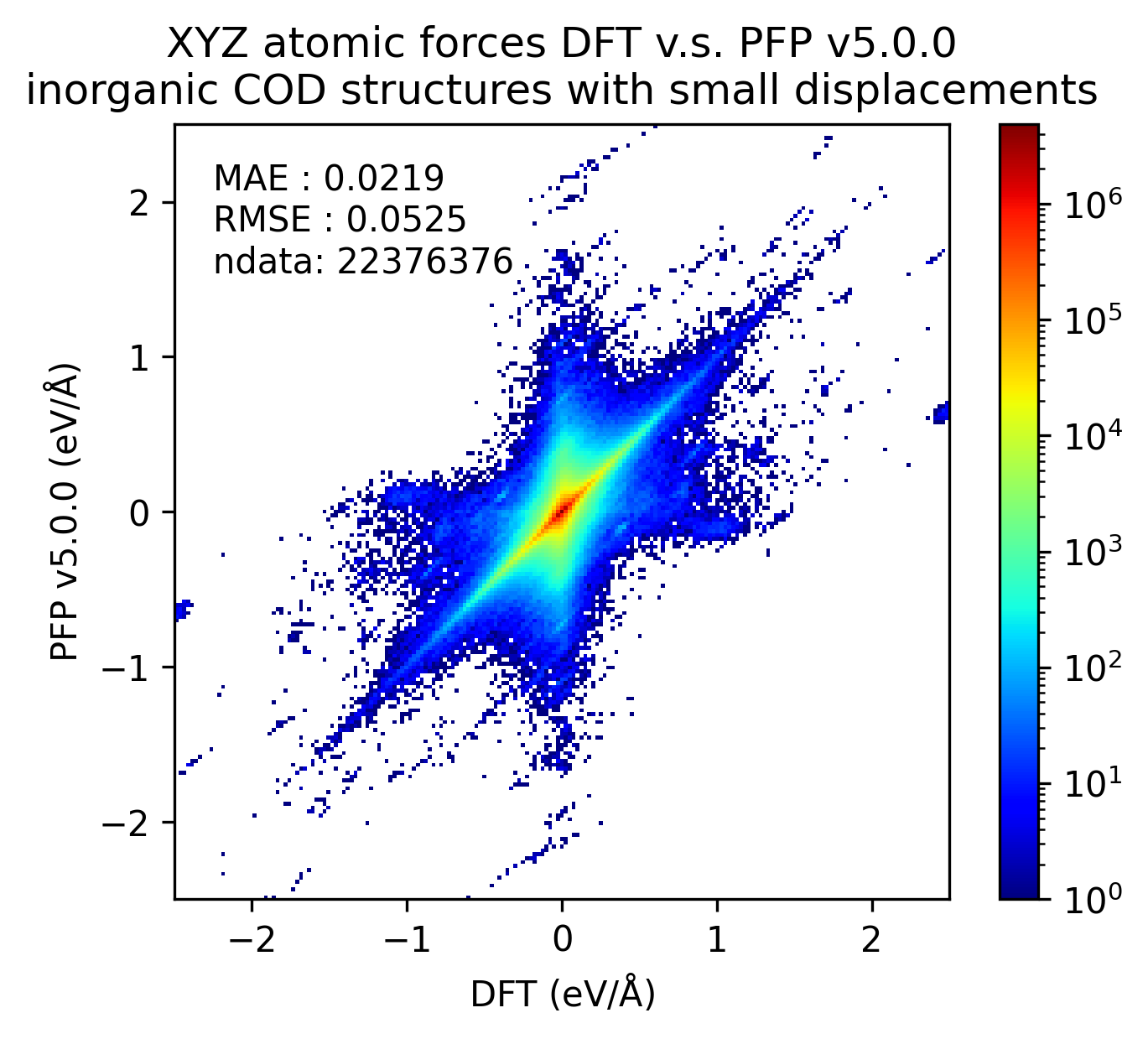 | 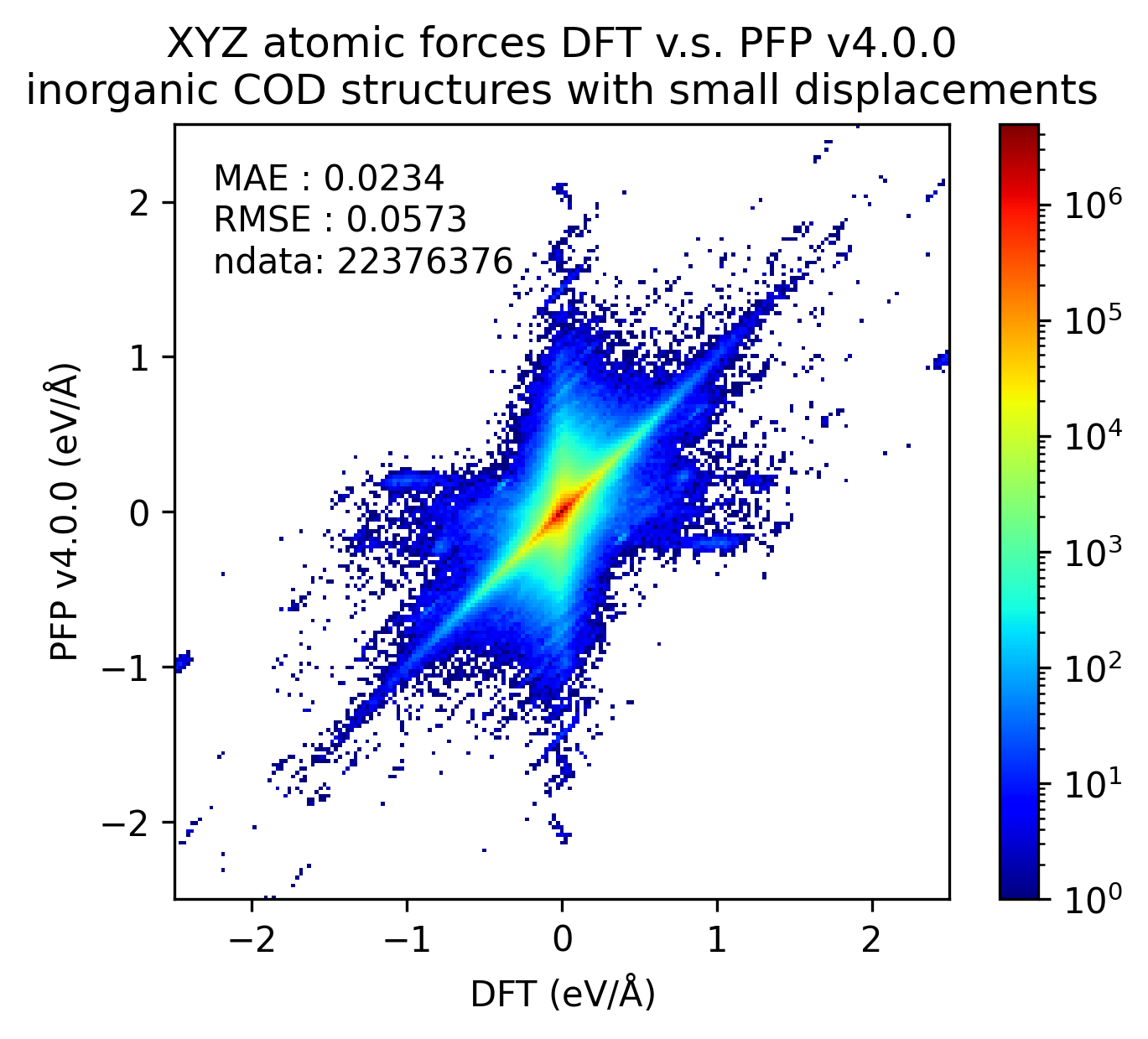 | 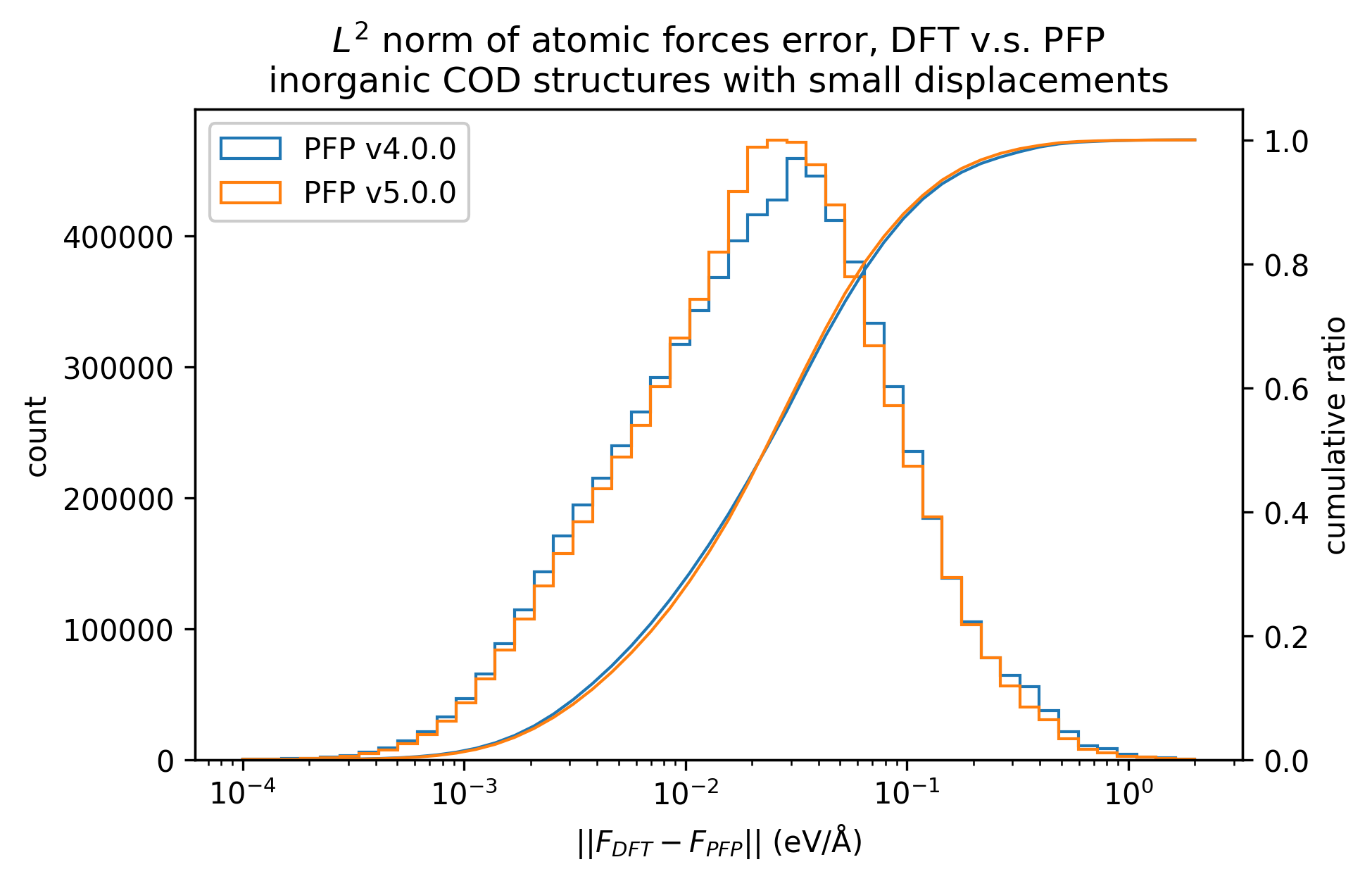 |
Based on these results, it can be said that PFP v5.0.0 has improved the reproducibility of energy and force, i.e., the reproducibility of potential energy surfaces, for organic and complex crystals compared to PFP v4.0.0. On the other hand, PFP v5.0.0 and v4.0.0 are comparable for inorganic crystals.
Acknowledgement
The latest version of Matlantis’s neural network potential, PFP v4, was developed using the National Institute of Advanced Industrial Science and Technology’s AI Bridging Cloud Infrastructure (ABCI) as well as PFN’s in-house supercomputers.
References
[1] Takamoto, So, et al. “Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements.” Nature Communications 13.1 (2022): 2991. https://www.nature.com/articles/s41467-022-30687-9
[2] “Matbench Discovery” https://matbench-discovery.materialsproject.org/
[3] “Materials Project” https://materialsproject.org/
[4] Wang, Hai-Chen, Silvana Botti, and Miguel AL Marques. “Predicting stable crystalline compounds using chemical similarity.” npj Computational Materials 7.1 (2021): 12. https://www.nature.com/articles/s41524-020-00481-6
[5] “Crystallography Open Database” https://www.crystallography.net/cod/
[6] Grimme, Stefan, et al. “A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu.” The Journal of chemical physics 132.15 (2010). https://pubs.aip.org/aip/jcp/article/132/15/154104/926936
[7] Grimme, Stefan, Stephan Ehrlich, and Lars Goerigk. “Effect of the damping function in dispersion corrected density functional theory.” Journal of computational chemistry 32.7 (2011): 1456-1465. https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.21759




