Unprecedented Speed for Materials InnovationThe AI simulation platform that accelerates new materials discovery
Matlantis transforms the research process for both computational and experimental chemists.
For computational chemists, it broadens application range and helps tackle complex challenges with greater speed and precision.
For experimental chemists, it provides instant access to predictive insights that guide and accelerate lab work.
By integrating simulation and experimentation, Matlantis enables a next-generation workflow powered by your expertise.


















Breaking the "Rules" of Conventional Simulation
Do these limitations sound familiar?
- "Calculations take too much time."
- "Large-scale systems
are tricky." - "When you want higher accuracy,
computational costs rise."
Matlantis Overturns That Premise
Unprecedented Calculation Speed
Instant inference on systems with tens of thousands of atoms
Simulate large-scale systems and long-term phenomena at previously unimaginable speeds, dramatically accelerating the trial-and-error cycle to deliver new discoveries in a fraction of the time.
True Versatility for
a Wide Variety of Materials
Perfect for a wide range of
materials and structures
Our platform supports materials discovery across all elements in nature, and is designed to simulate a wide variety of structures—from bulk crystals and surfaces to interfaces and amorphous systems—offering powerful support for the search for unknown materials.
AI Models with
Quantum-Mechanical Accuracy
No data preparation or additional learning required
Our software features state-of-the-art AI models, pre-trained on a meticulously curated quantum mechanical dataset. Predict physical properties with high accuracy from the start—no specialized knowledge or additional training needed.
Secure and
Hassle-Free Computation in the Cloud
No infrastructure setup
or management required
Access our platform instantly from any web browser—no environment to build or hardware to maintain. We manage all infrastructure, delivering a seamless, zero-maintenance experience so you can focus entirely on research and development.
Functions and Features
Accelerating R&D Enables Next-Generation Materials Discovery
Matlantis, equipped with a proprietary AI model, can perform simulations tens of millions of times faster than conventional electronic structure methods while maintaining high accuracy. Supporting a total of 96 elements—including all that occur naturally—Matlantis enables complex calculations that were not previously possible. By predicting atomic-level phenomena that cannot be captured by experiments alone, Matlantis contributes to new material design, advanced property prediction, and shortened development cycles.
Features Tailored to Each User
Matlantis
Portable Guide
Everything You Need to Know — Features, Benefits, and More

Powered by World-Class AI Technology
Machine learning interatomic potentials: AI models that capture atomic behavior
We set out to create a general-purpose machine learning interatomic potential capable of predicting the properties of any material; once considered "too difficult to achieve," we have now succeeded.
Using cutting edge AI technology, we have developed a unique model called the PFP (PreFerred Potential).
Core Technology
What is our AI Model, PFP?
Introducing PFP, a unique AI model that combines high speed, accuracy, and versatility.
Latest Version and History
PFP has undergone multiple performance upgrades since its launch. Learn about the latest versions capabilities.
Verification of Predicted Performance of PFP
Learn more about the accuracy of PFP compared to DFT.
Comparison with Open Source MLIP
This page presents a comparison between PFP and other open-source machine learning interatomic potentials (MLIPs).
PFP-Based Applied Technology
LightPFP
For users who require even larger-scale calculations, we’re introducing a feature that allows you to build custom potentials using PFP.
ReactionString/RestScan
We’re introducing two features: ReactionString, for optimizing reaction paths, and RestScan, for generating new ones.
GRRM20 with Matlantis
We’re introducing features for comprehensive reaction path searches and the construction of complex reaction networks.
Matlantis CSP
We will introduce a function that quickly and automatically discovers stable crystal structures by simply providing a combination of elements.
PFP Descriptors
This feature provides access to descriptors generated by PFP, which can be used for physical property prediction and other machine learning tasks.
Calculation Cases
Matlantis is applicable across a wide range of areas explored in modern materials science, including energy, electronics and polymer chemistry.
Pharmaceuticals
High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
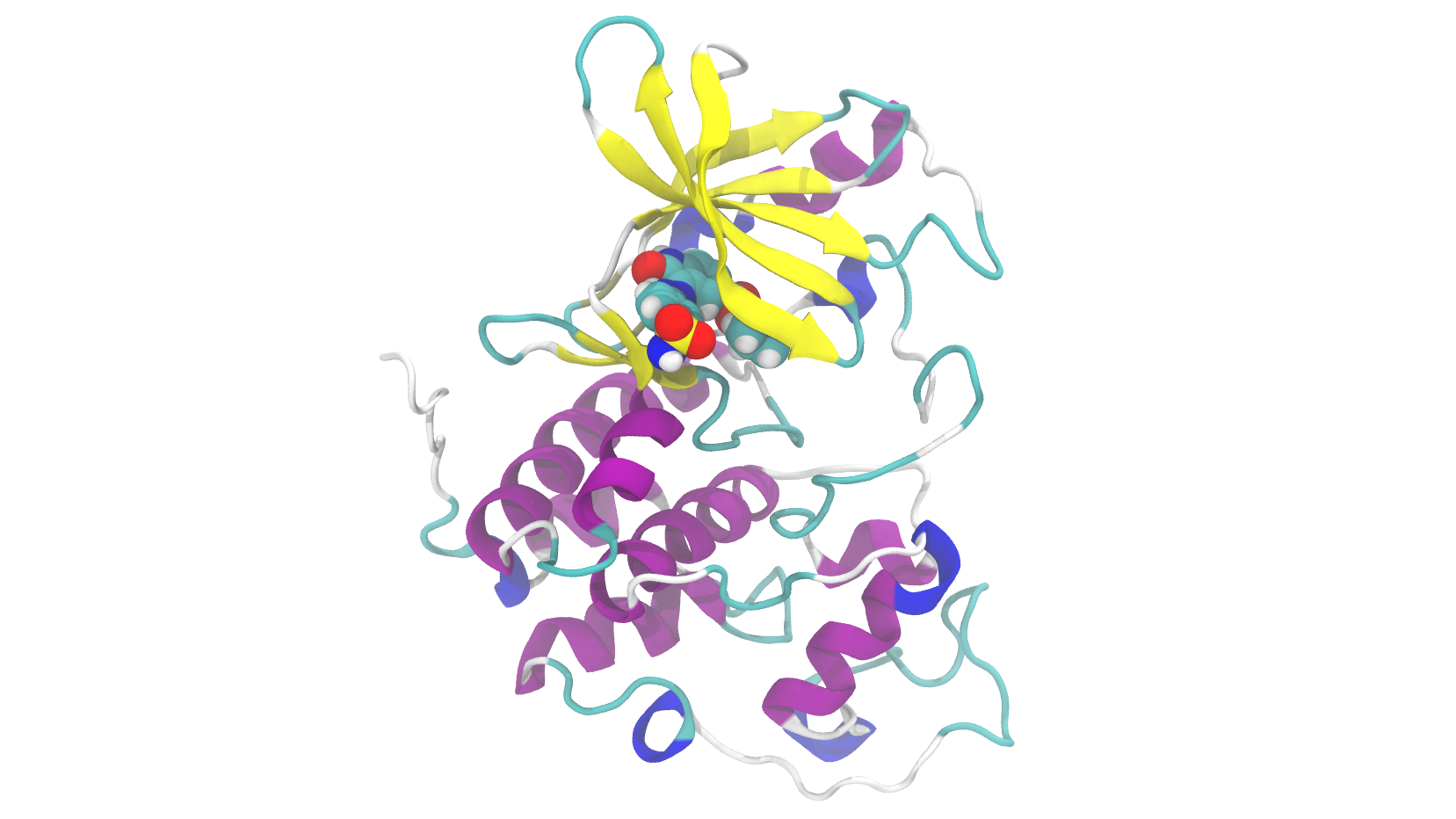
screening
Materials Informatics
Semiconductors
Data-Driven Method for Discovering Low Adsorption Energy Molecules on Si Surfaces
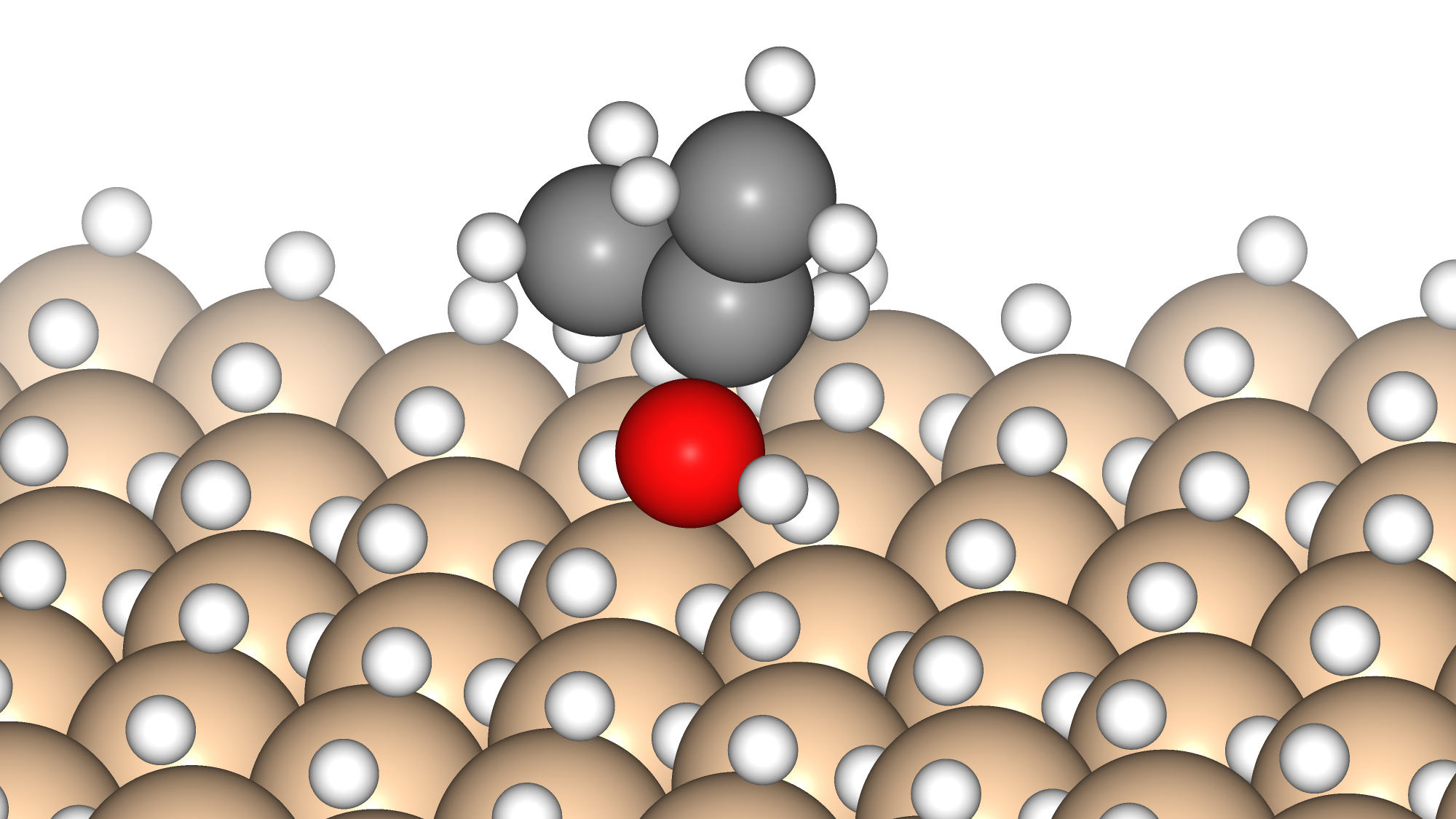
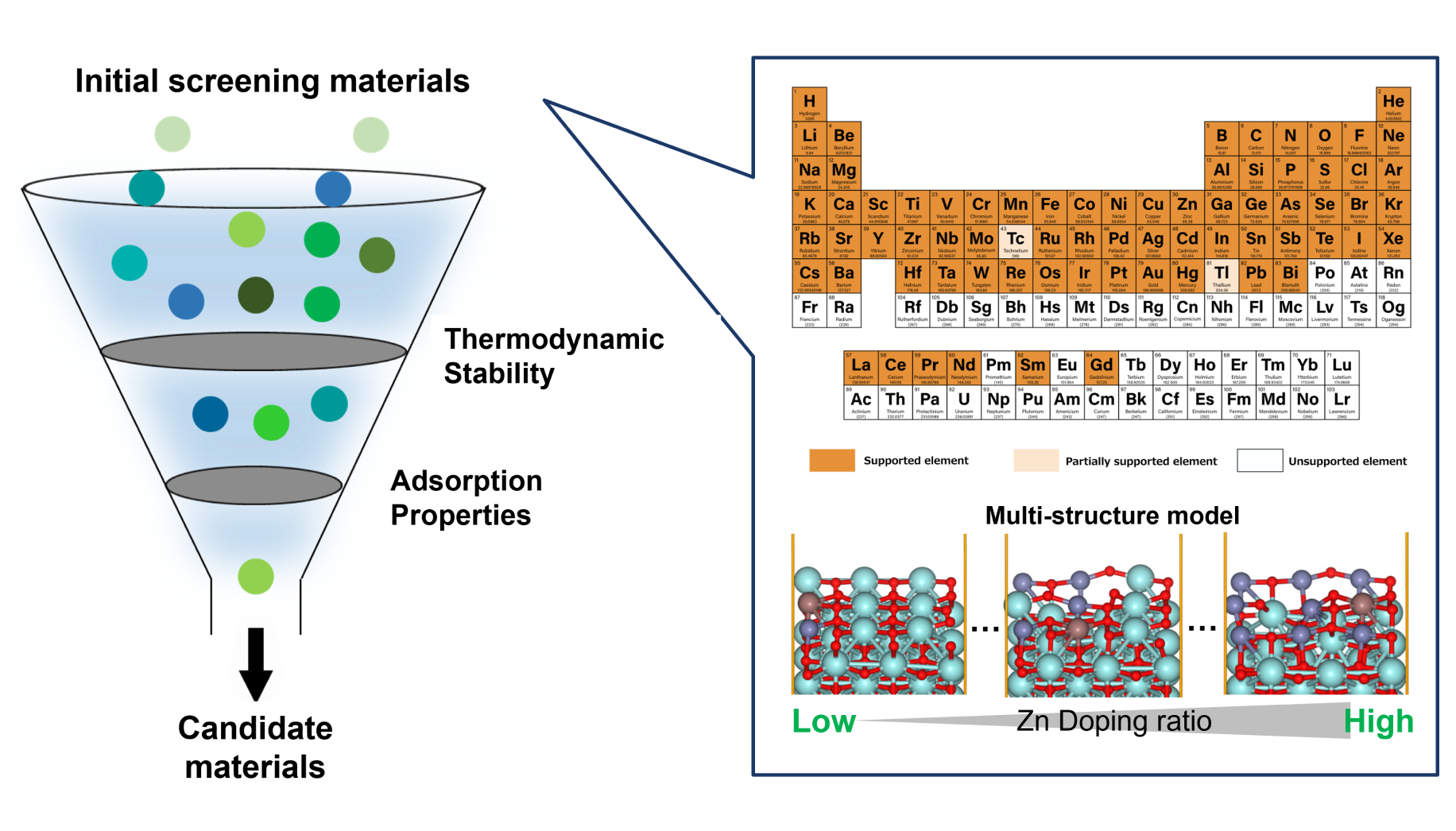
Methanol synthesis catalysts
Large-scale screening
Calculation and Verification of Methanol Synthesis Catalysts
Atomic displacement parameter (ADP)
thermoelectric materials
Direct derivation of atomic displacement parameters using NNP-MD

Ceramics
High Entropy Materials
electrolytic
A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides

Ready to unlock your next discovery? Let's explore how Matlantis can transform your research.
Trusted by Industry and Academia
Matlantis has a proven track record helping customers across a range of industries with their material exploration.
Corporate and Research Institute Customers
150+
We support materials discovery at over 150 companies and research institutes worldwide.
Matlantis Users
900+
Matlantis is used not only by computational chemists but also by experimental researchers.
Number of Research Papers Using Matlantis
100+
Since starting operations in 2021, we have built a solid track record in materials science.
Information as of March 2026
It's really amazing that you can get results just by giving it a go without having to narrow down the phenomenon.

What used to take about three months was shortened to just one week by using Matlantis.
There's a reason we are chosen by cutting-edge sites.
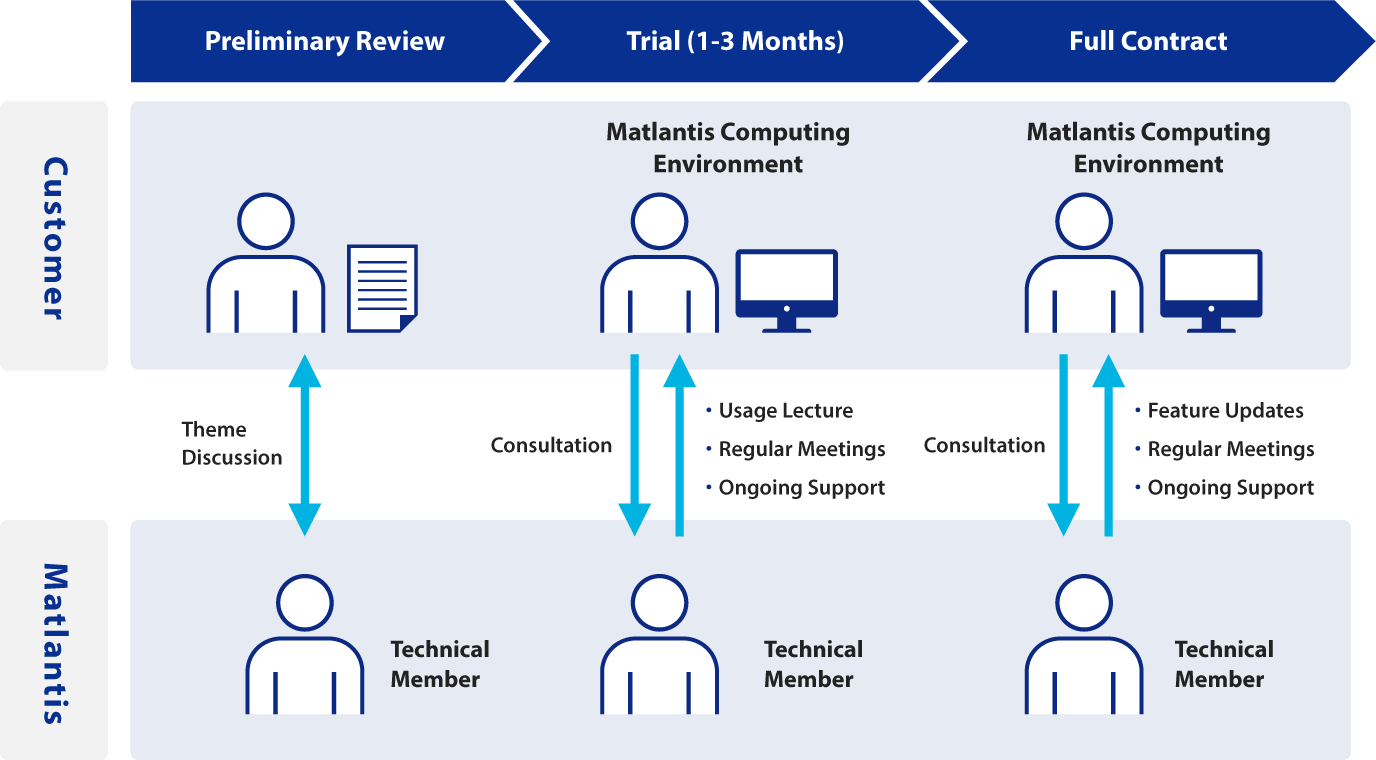
Environment and Onboarding Process
Matlantis provides an optimal environment tailored to your needs,
along with comprehensive support to ensure long-term, successful use.

Matlantis is a Step towards Changing Material Development
-




 URL
URL
Copied