Cases
We've highlighted some of the most notable examples from user interviews, simulation cases, and research papers. Browse by category to explore the details that interest you.
Featured Cases
Featured Customer Stories
It was impossible to predict based on past experience or intuition.
Creating new combinations and encounters
Kuraray Co., Ltd.Chemicals
Combined with AI and experimental data, computational chemistry contributes to raising the level of research and development. “We expect that it can be a powerful tool to compete globally.”
Learn More
From left to right: Dr. Kamata, Senior Manager of the Planning and Administration Department And Digital Solution Department, Research and Development Division, Mr. Sugoh, General Manager of the Research and Development Division, and Dr. Miura, Manager of the Digital Solution Department established within the Research and Development Division
Featured Calculation Examples
High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
Pharmaceuticals
概要 創薬研究において、リード最適化段階でのタンパク質-リガンド結合親和性の正確な予測は、化合物選択の成否を左右します。しかし、量子化学計算による高精度な相互作用計算は1構造あたり数十時間を要し、大規模な化合物ライブラリへの適用は現実
Learn More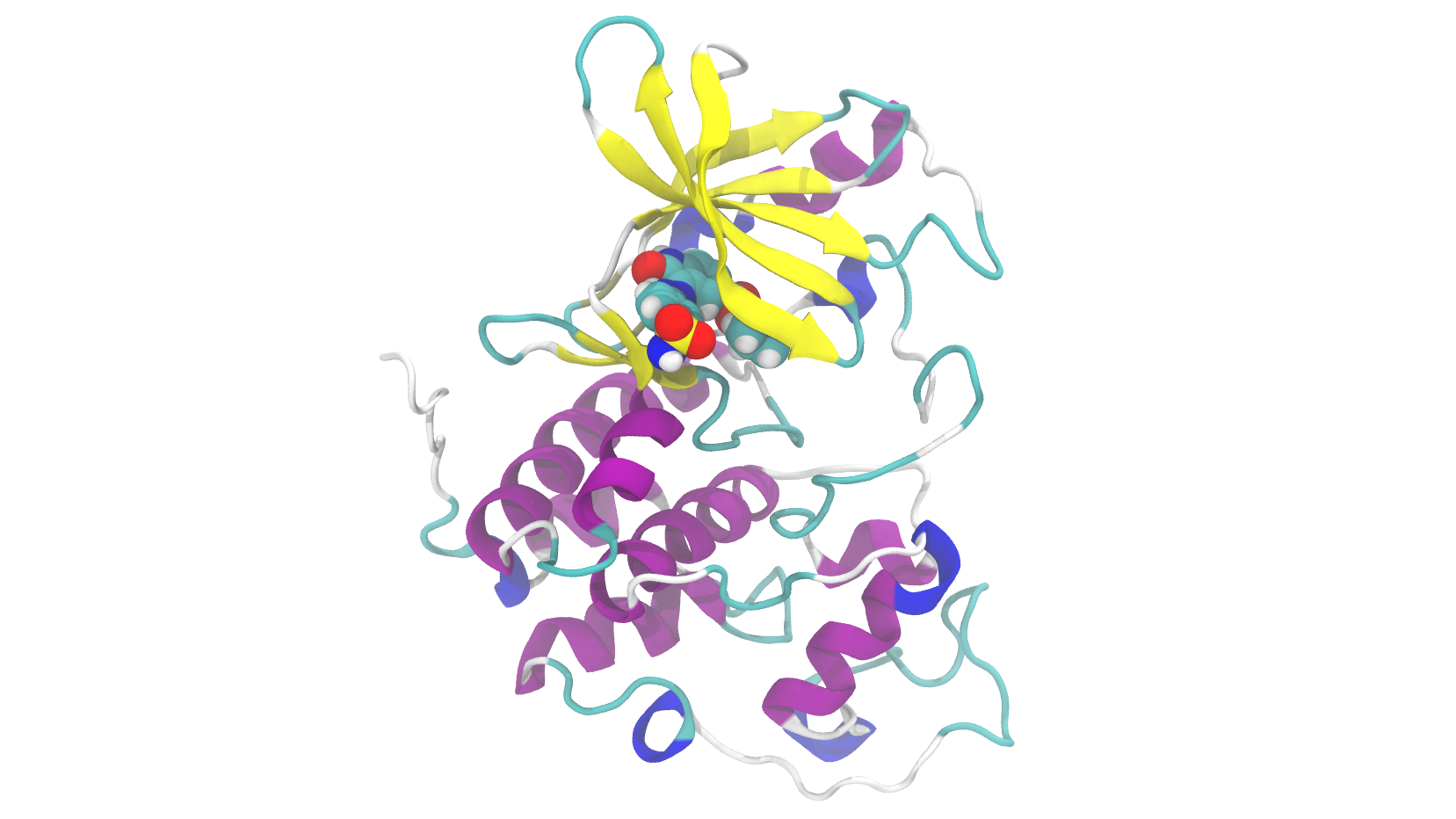
Featured Published Papers
A statistical understanding of oxygen vacancies in distorted high-entropy perovskite oxides
Matlantisを用いた論文
セラミックス
High-entropy perovskite oxides have emerged as promising electrode materials for solid oxide electrolyzers. However, their compositional complexity makes the formation of oxygen vacancies, which influence properties such as oxygen ionic conductivity and thermal expansion, challenging to predict. Here, we experimentally measure changes in oxygen vacancy concentration for fourteen perovskite oxides with high and low-entropy A-site compositions, finding a dependence on cation size variance in addition to divalent cation fraction. Atomistic simulations using…
Search for cases by category
List of customer cases
We will introduce the background, challenges, usage methods, and results obtained by companies that have introduced Matlantis. Through real voices from the field, we will provide you with hints that will be useful when considering introducing Matlantis.
List of calculation examples
We will introduce specific simulation examples using Matlantis for various material systems. You can see how high speed and high accuracy calculations are achieved.
List of published papers
This site mainly features papers published by Matlantis users about research using Matlantis, as well as papers about machine learning potential and PFP, which are core technologies of Matlantis.
How to cite
For information on how to cite Matlantis papers and case studies, please see the link below.
Follow us on social media to get timely updates on new case studies and more!
Beyond Human Intuition — with Matlantis
Matlantis
Portable Guide
Everything You Need to Know — Features, Benefits, and More

-




 URL
URL
Copied