Reaction analysis of TMA in a system that mimics the surface of silicon oxide film
Tokyo Electron Limited

Overview
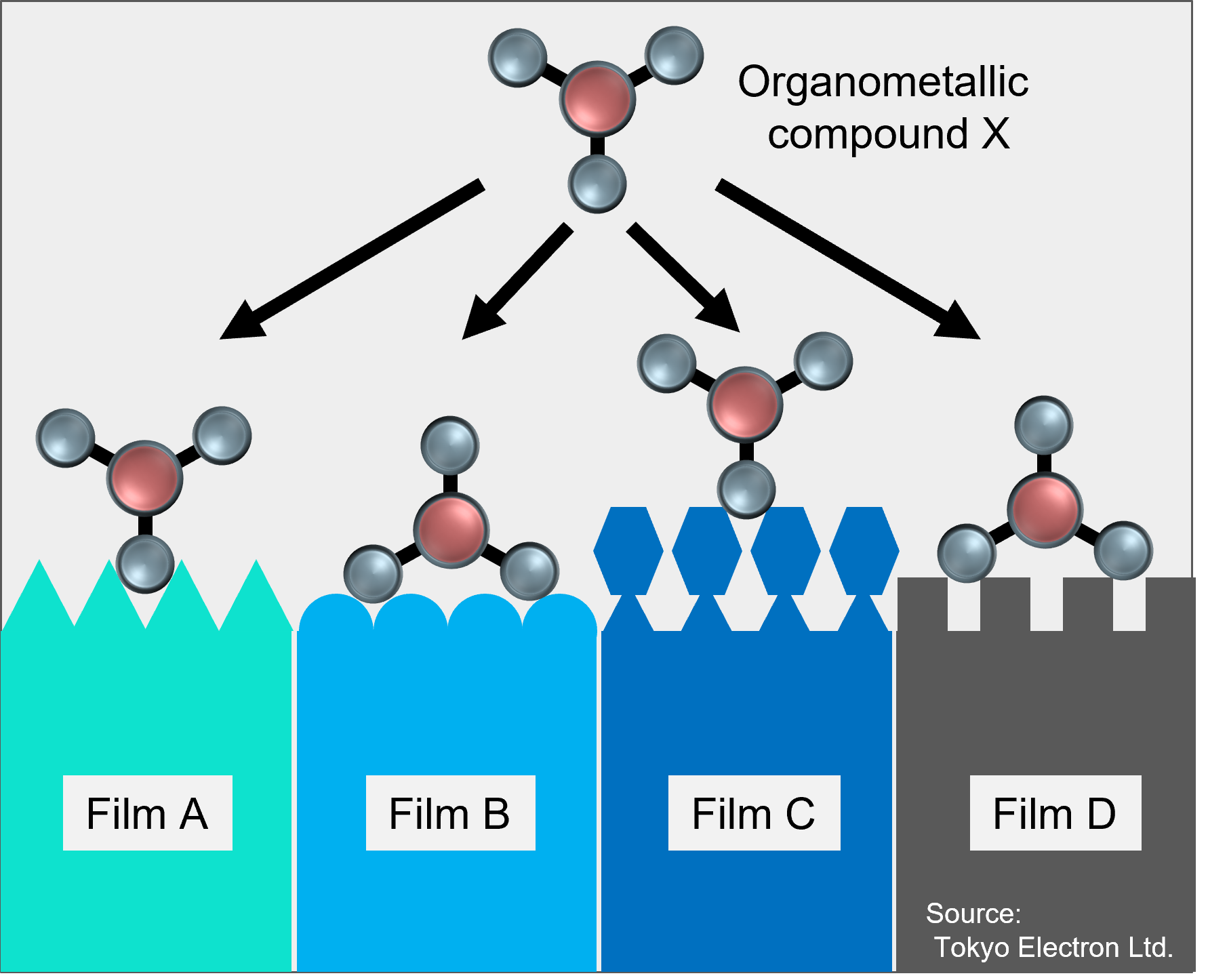
Adsorption of organometallic compounds on semiconductor and silica gel surfaces is an important step in the series of reactions involved in thin film deposition and organocatalyst synthesis [1,2].
Physicochemical analysis of the reaction mechanism can be useful in selecting the best source molecules, but it has not been realistic in terms of computational resources to analyze the reaction mechanism on various solid surfaces depending on the crystalline phase.
Calculation models and methods
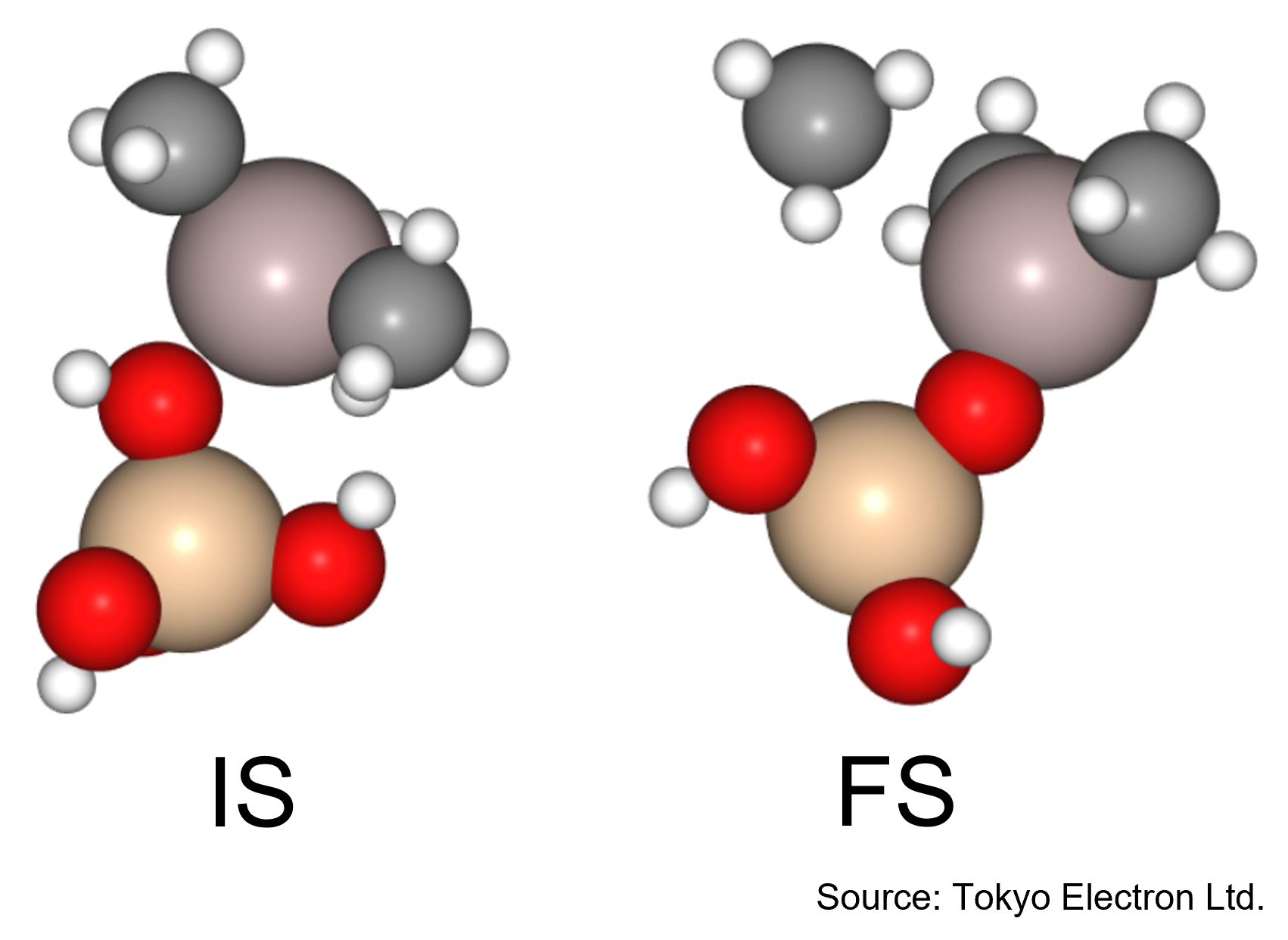
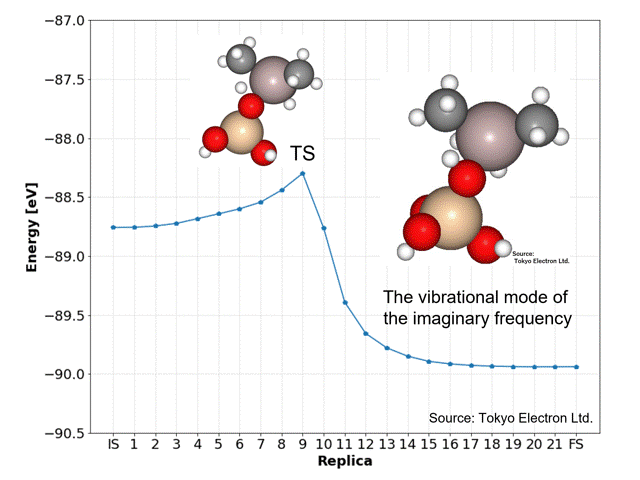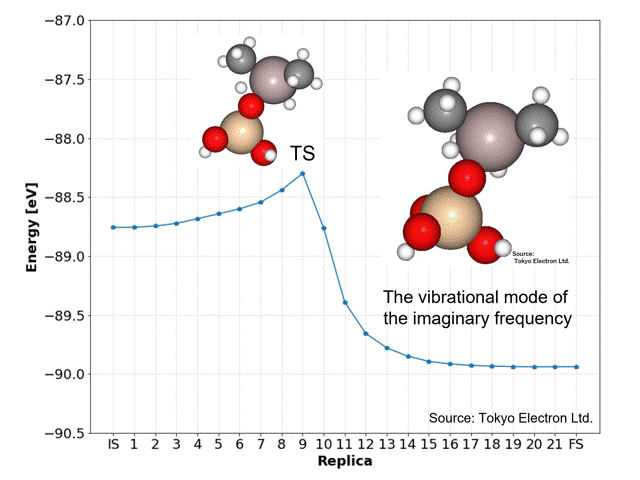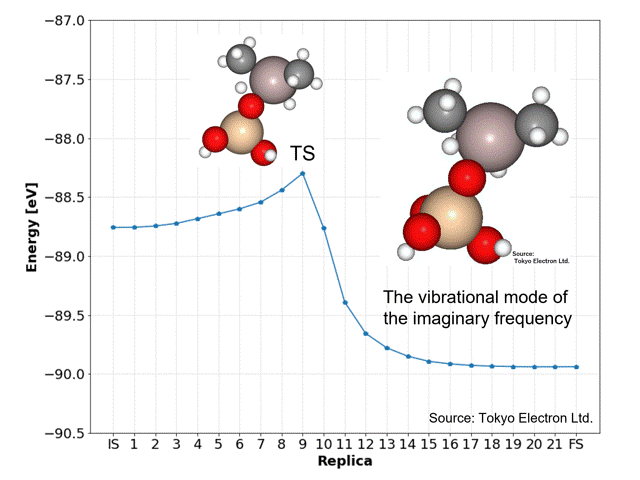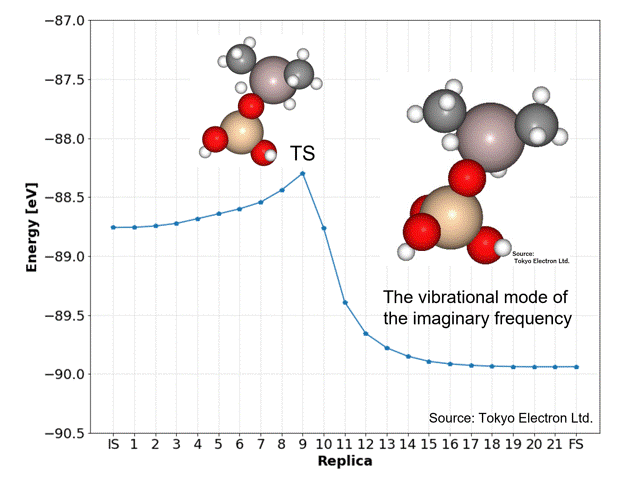
In this study, we focused on the reaction in which the silanol group of orthosilicate (OSA) adsorbs on trimethylaluminum (TMA) to form an Al-O bond and methane is desorbed.
In the initial state (IS), TMA is placed in the opposite direction of the proton of the silanol group of OSA, and in the final state (FS), the product has an Al-O bond and methane attached by the proton desorbed from the silanol group.
NEB calculations were performed using these structures.
Calculation Results and Discussion
NEB calculations yielded dissociation activation energies with similar accuracy to the DFT calculated data.
The ninth replica structure was predicted to be the transition state (TS), which was also predicted to be a reasonable structure from the vibrational frequency and vibrational mode calculations. The IRC calculations also predicted the IS and FS structures to be reasonable.
The NEB calculations were performed using the idpp approach with a maximum force on all atoms of 0.01 [eV/Å] and a spring constant k of 0.1, using the D3 correction (CRYSTAL_PLUS_D3, Ver.1.1.0) for PFP, and took 769 iterations and 21 minutes and 7 seconds.
A similar calculation using the DFT approach with a typical calculation cluster would take several days to several weeks of calculation time (including queue time and run time). The ability of Matlantis to complete the screening by NEB calculation for a system with multiple possible starting and ending states in about one day has the potential to greatly accelerate the resolution of issues through simulation.
Calculation Condition