テーマ概要
半導体やシリカゲル表面への有機金属化合物の吸着は、成膜や有機触媒合成に関わる一連の反応において重要なステップとなります[1,2]。
その反応機構に関する物理化学的な解析は、最適な原料分子の選択に役立てることができますが、結晶相に応じたさまざまな固体表面で反応機構を解析することは、計算資源的に現実的ではありませんでした。
計算モデルと計算方法
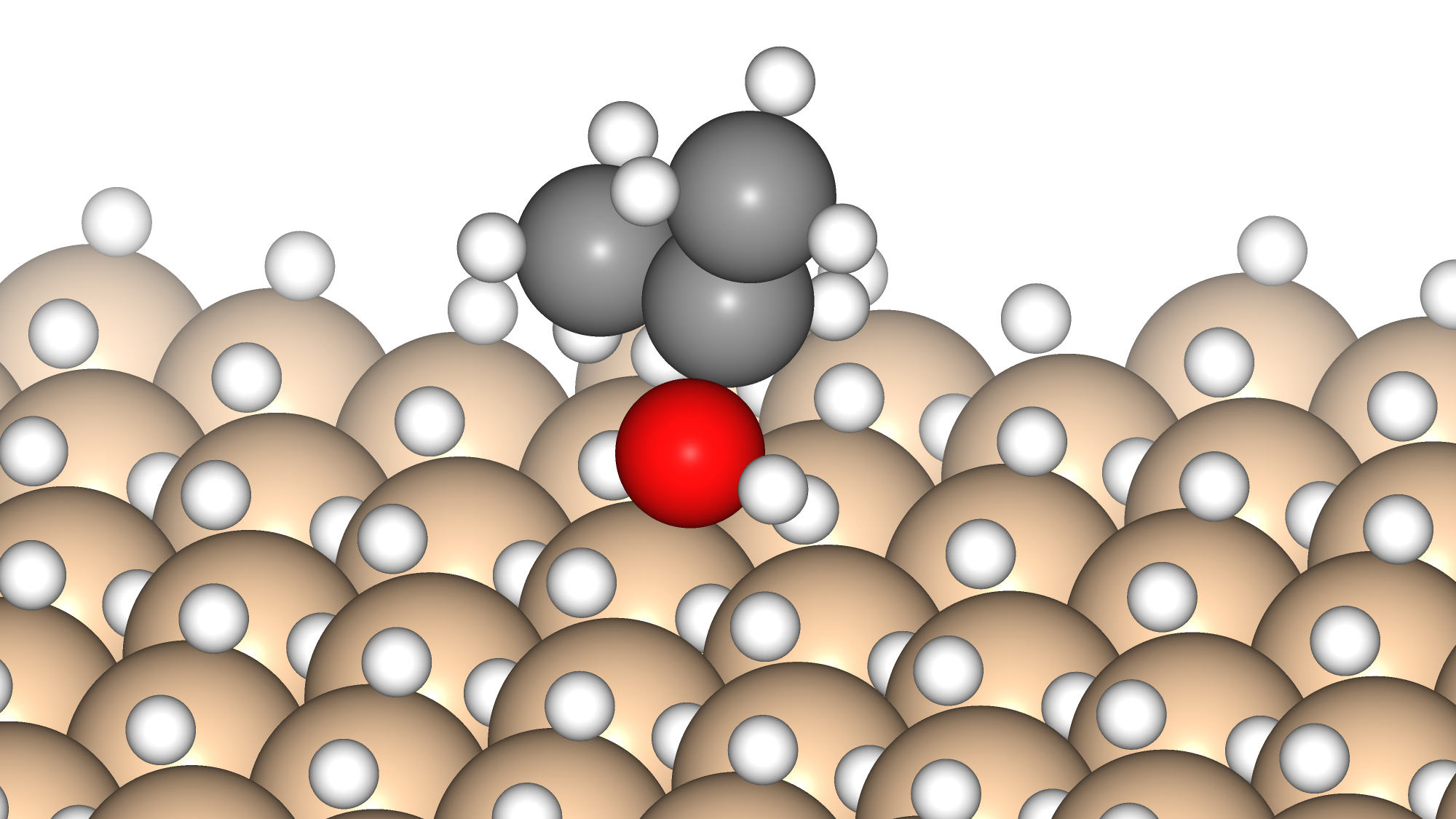
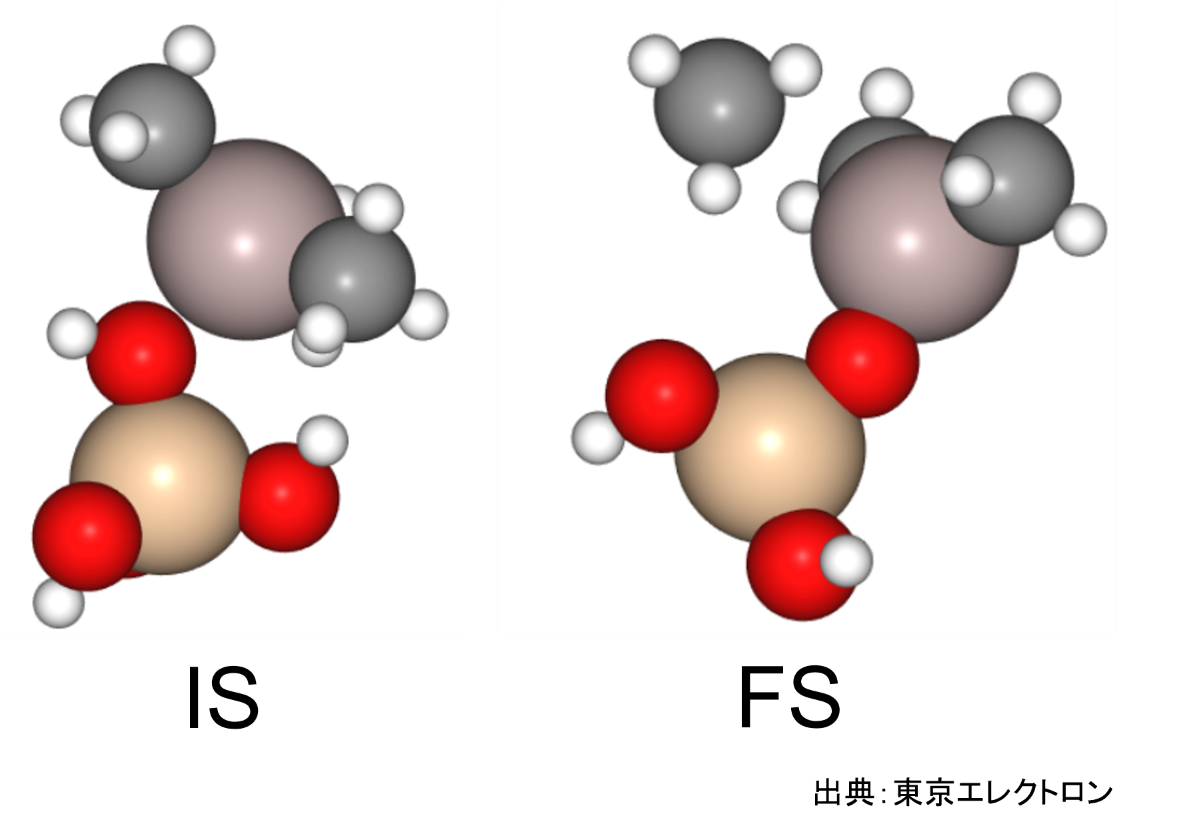
今回はオルトケイ酸(OSA)のシラノール基がトリメチルアルミニウム(TMA)に吸着してAl-O結合を形成し、メタンが脱離する反応を対象としました。
始状態(IS)ではOSAのシラノール基のプロトンの反対方向にTMAを配置し、終状態(FS)としては、Al-O結合を有する生成物と、シラノール基から脱離したプロトンが付加したメタンとしました。
これらの構造を用いてNEB計算を実施しました。
計算結果と展望
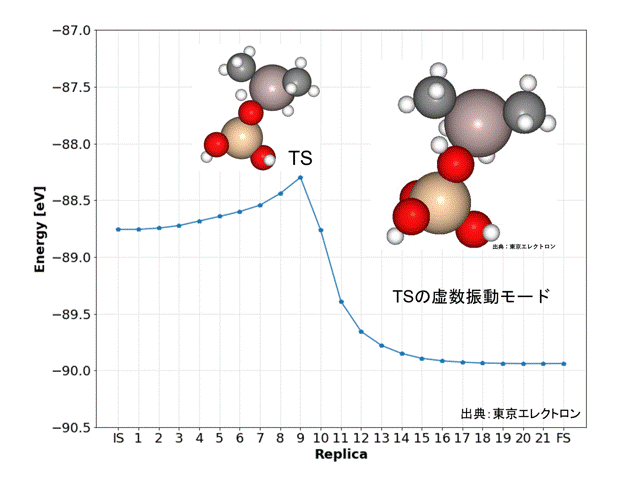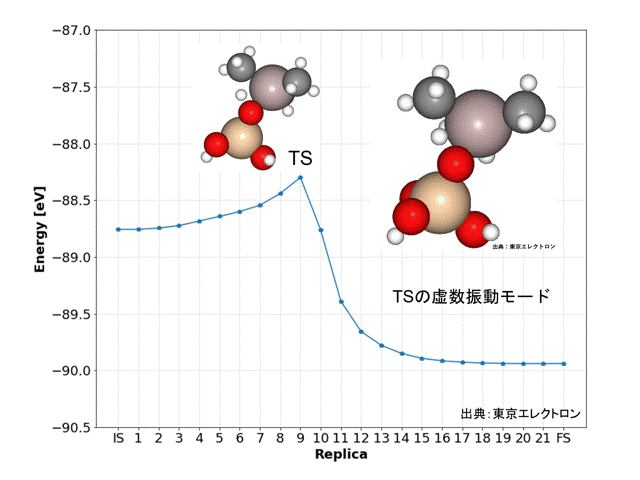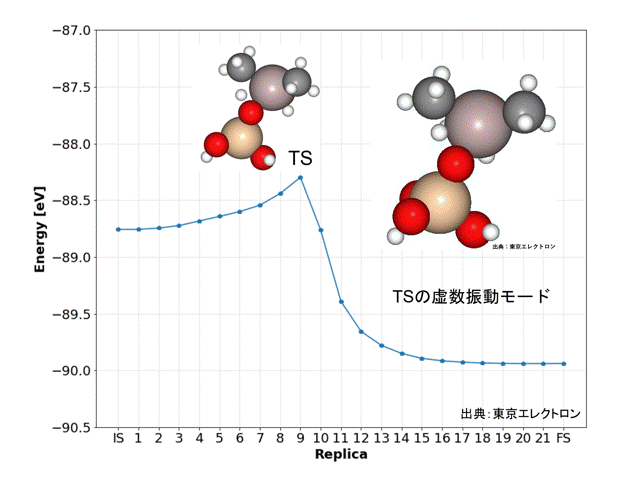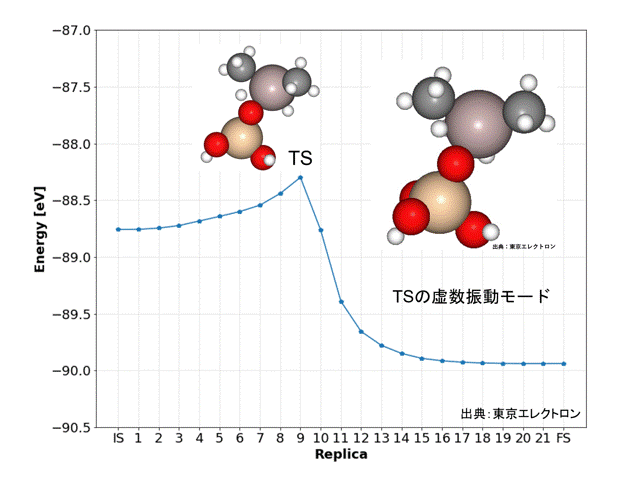
NEB計算により、DFT計算データと同様な精度で解離活性化エネルギーを得ることができました。
Replicaの9番目の構造が遷移状態(TS)と予想され、振動数および振動モードの計算からも、妥当な構造だと予想されました。また、IRC計算の結果からもISとFSの構造が妥当だと予想されました。
今回のNEB計算では全ての原子に働く力の最大値を0.01 [eV/Å]、ばね定数kは0.1として、PFPのD3補正(CRYSTAL_PLUS_D3, Ver.1.1.0)を用いてidppアプローチで計算しましたが、769回のiterationで21分7秒の計算時間で終了しました。
同様な計算を一般的な計算クラスターを用いてDFT法で計算した場合、 キュータイムも含めると数日から数週間の計算時間を要すると考えられます。複数の始状態・終状態の想定される系でのNEB計算によるスクリーニングをMatlantisで1日程度で完了できることは、シミュレーションによる課題解決を大きく早められる可能性を秘めています。
計算条件

参考文献
[1] Organometallics 2001, 20(16), 3519–3530 https://doi.org/10.1021/om0102596 [2] Macromol. Chem. Phys. 2000, 201, 1334-1344 https://onlinelibrary.wiley.com/doi/10.1002/1521-3935%2820000801%29201%3A12%3C1334%3A%3AAID-MACP1334%3E3.0.CO%3B2-%23 [3] J. Phys. Chem. C 2015, 119(32), 18380–18388 https://doi.org/10.1021/acs.jpcc.5b05261事例提供者プロフィール
東京エレクトロン株式会社

タグ
本事例の公開日:2022.05.09
-




 URLを
URLを
コピーしました