Monolayer to bilayer phase transformation of CaSi2Fx(0<x<1.5)
Introduction
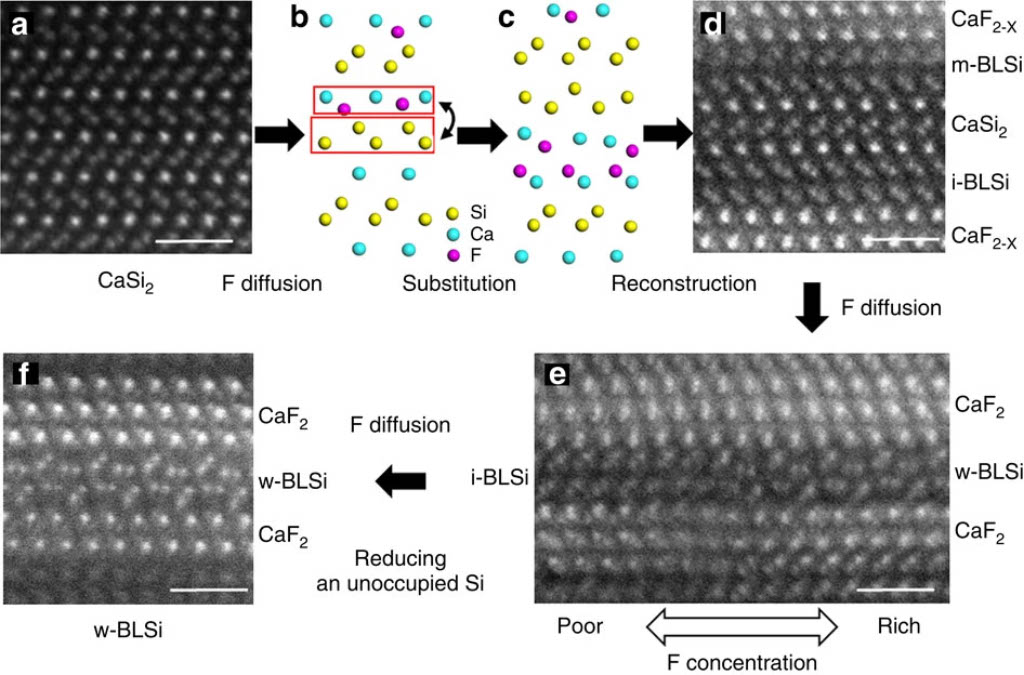
CaSi2 is used for fabricating silicene and silicene derivatives[1]. The crystal structure of CaSi2 consists of alternatively stacked structures of Ca and Si monoatomic layers. The crystal structure transforms to bi-layered and multi-layered structures by fluorine diffusion.
These diffusion and intercalation of atoms into the layered materials are used to control the structure of 2D materials, exfoliations, and electrode materials. The phase stability with respect to the atomic diffusions is typically studied using DFT simulations and cluster expansion[2].
In this use case, we reproduced the DFT result in literature, and further calculate the stability of other polymolphs. Our results show that the phase transformation of CaSi2Fx agrees well with the experiments, and large-scale MD simulations with over a thousand atoms provide insight into the reaction dynamics.
Computational details
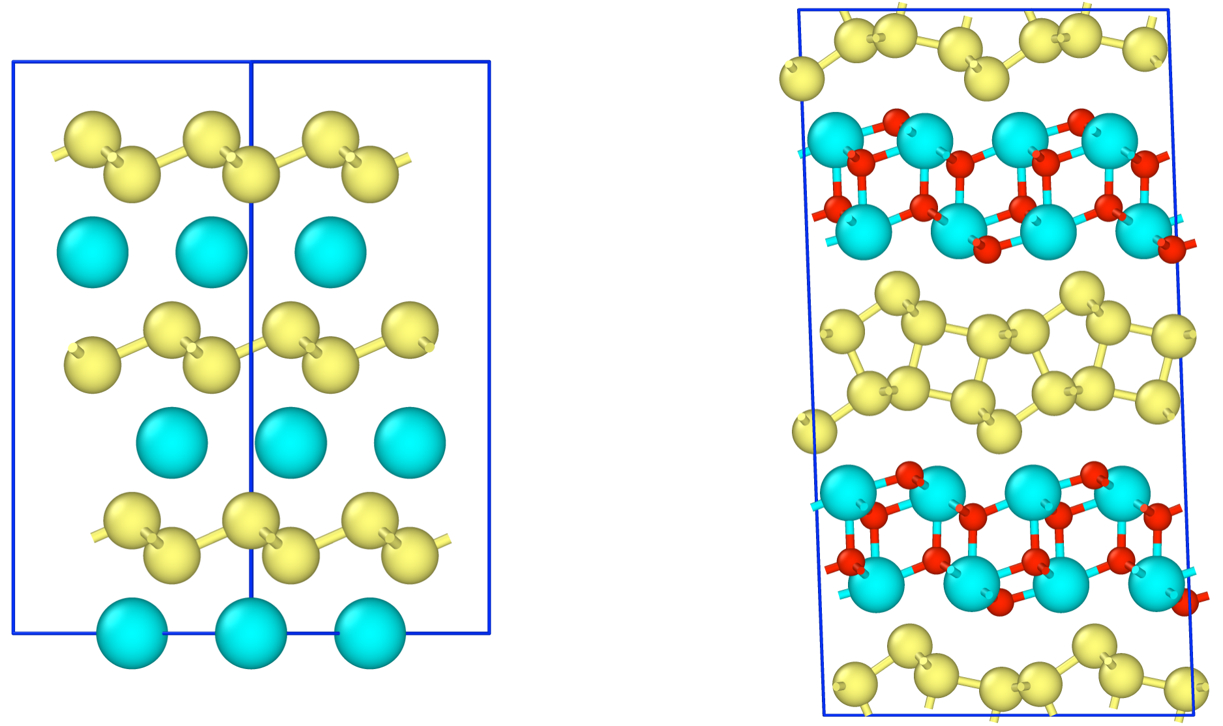
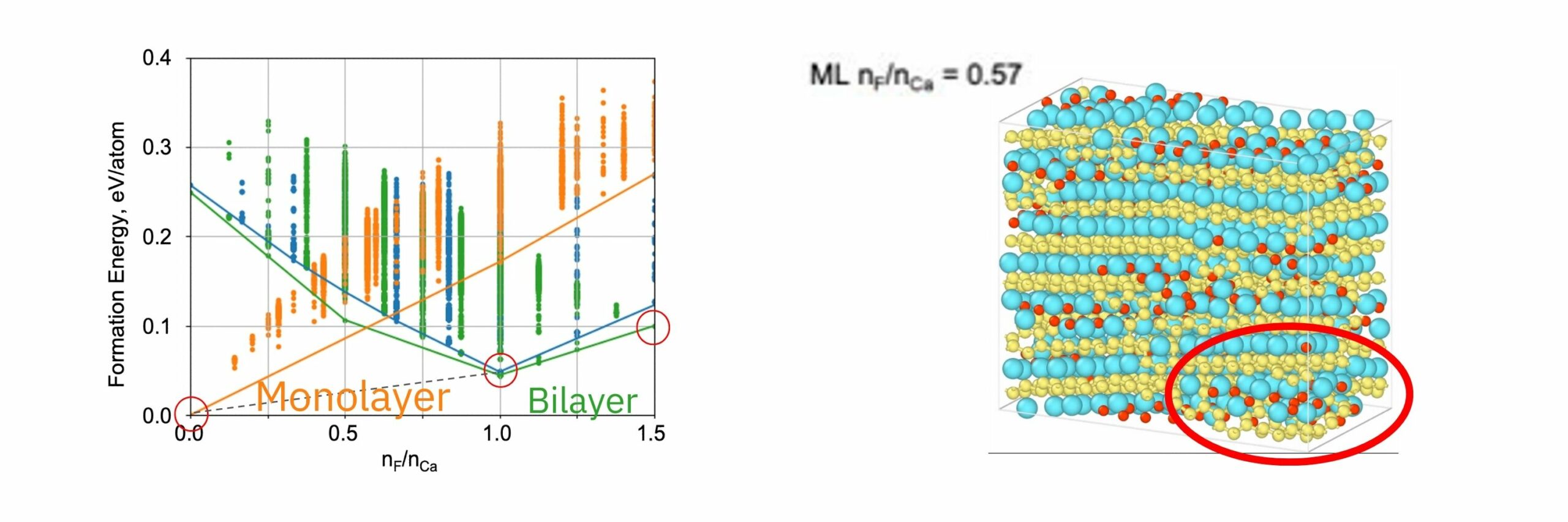
The formation energy of CaSi2Fx(0<=x<=1.5) was calculated using PFP with respect to the Ca/F composition ratio x. Three structural models of CaSi2Fx, monolayer, i-bilayer, and wavy-bilayer, were used as the host materials, and the stable fluorine configurations were examined using icet package[3].
Results and Discussions
The energy convex hull consists of monolayer, i-bilayer and wavy-bilayer structures at x=0.0, 1.0, and 1.5, respectively. The bilayer structure is the most stable when F/Ca ratio exceeds 0.5, in quantitative agreement with STEM observations.
In the MD simulations, formation of bilayer structure of CaSi2 was observed at high fluorine density region.
This use case demonstrates that the PFP accurately reproduces the experimental results and can be applied to the analysis of phase transformation due to ion diffusion.
Computational conditions