層状材料CaSi2Fx (0<x <1.5)の相変態
テーマ概要
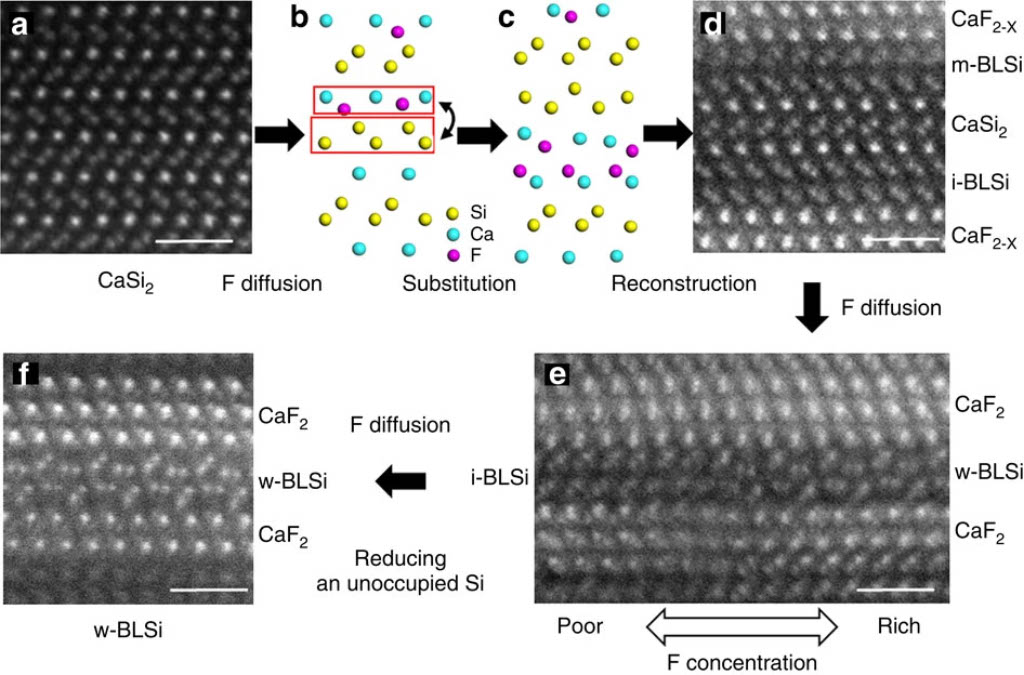
層状材料CaSi2は単原子層のCaとSiとが交互に重なった構造であり、シリセン化合物の原料として用いられる[1]。CaSi2にFを拡散させると単原子層の構造から2原子層、3原子層の構造への構造相転移が起こる。
このような層状材料への原子挿入に伴う反応は、シート状材料の剥離や制御の他、2次電池の電極材料などで重要である。一方、組成に対して構造安定性を調べる必要があり、従来は第一原理計算の結果に対してクラスタ展開でフィッティングすることで計算が行われていた[2]。
この事例では、DFTの文献結果をPFPで再現するとともに、文献にない構造についても安定性の評価を行った。その結果、実験と対応する構造相転移が計算から確認できた。さらに1000原子以上のMD計算を行うことで、反応のダイナミクスについても解析を行った。
計算モデルと計算方法
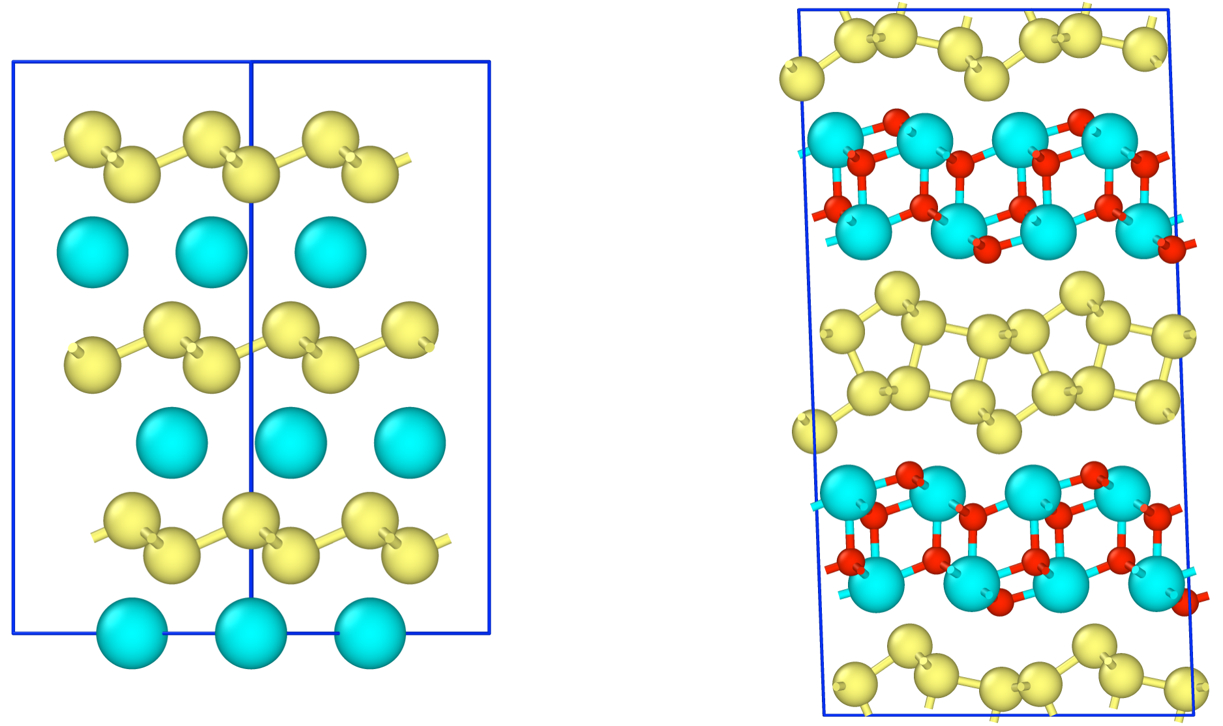
CaSi2Fx(0<=x<=1.5) について、組成比xに対して形成エネルギーを計算し、安定構造を求めた。構造モデルには、単原子層のCaSi2の他に、実験で観測された2原子層の構造(i-bilayer、wavy-bilayer)について計算を行った。
安定なF配置の探索にはpythonパッケージicetを用いた[3]。icetで生成した様々なF配置の構造全てについて、PFPで形成エネルギーを評価した。
計算結果と展望
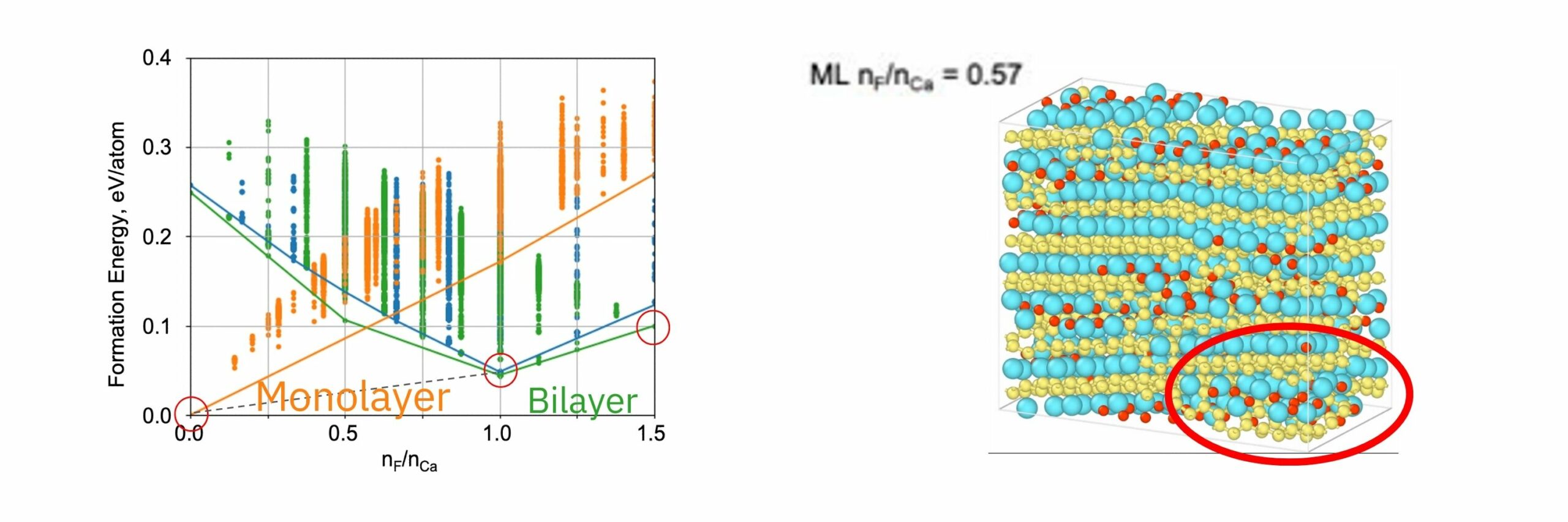
形成エネルギー計算の結果、F/Ca比 0.5以上で単原子層構造よりも二原子層構造が安定となるというDFTの文献と一致する結果が得られた。F/Ca比1.0でi-bilayer構造、1.5でwavy-bilayer構造が安定となる。計算結果は、STEMで観測された構造相転移の結果と定量的に一致した。
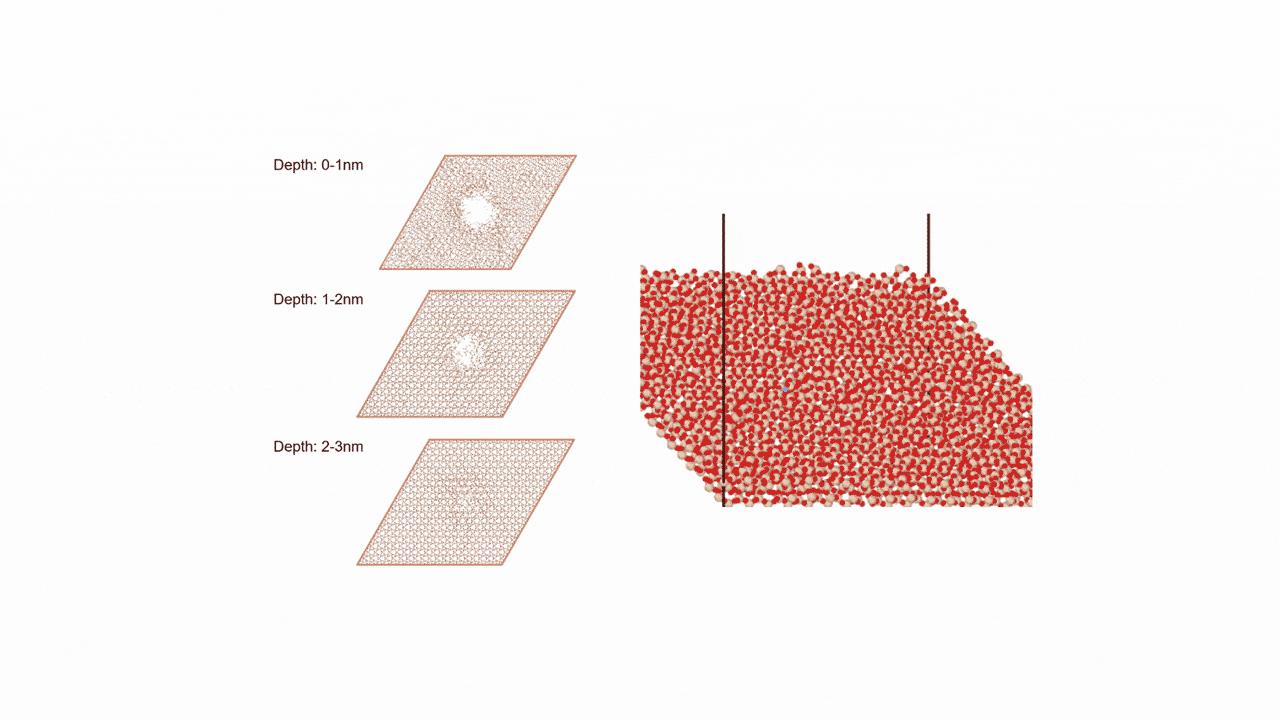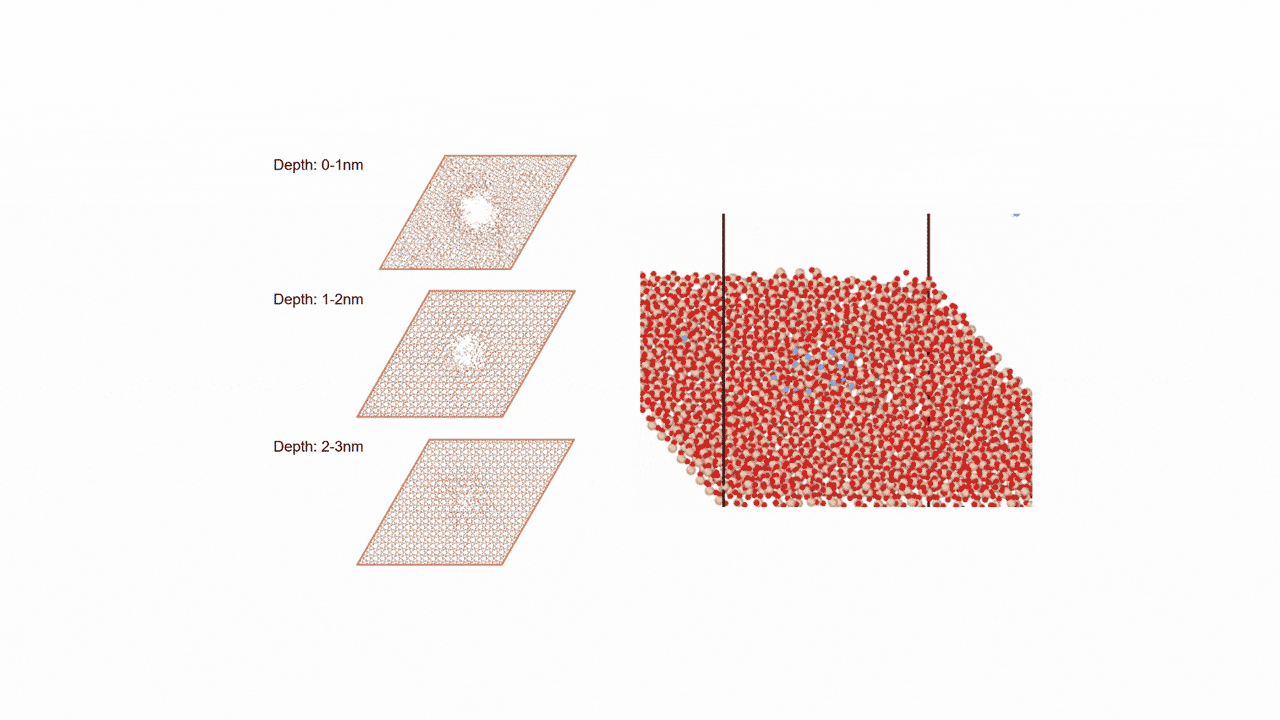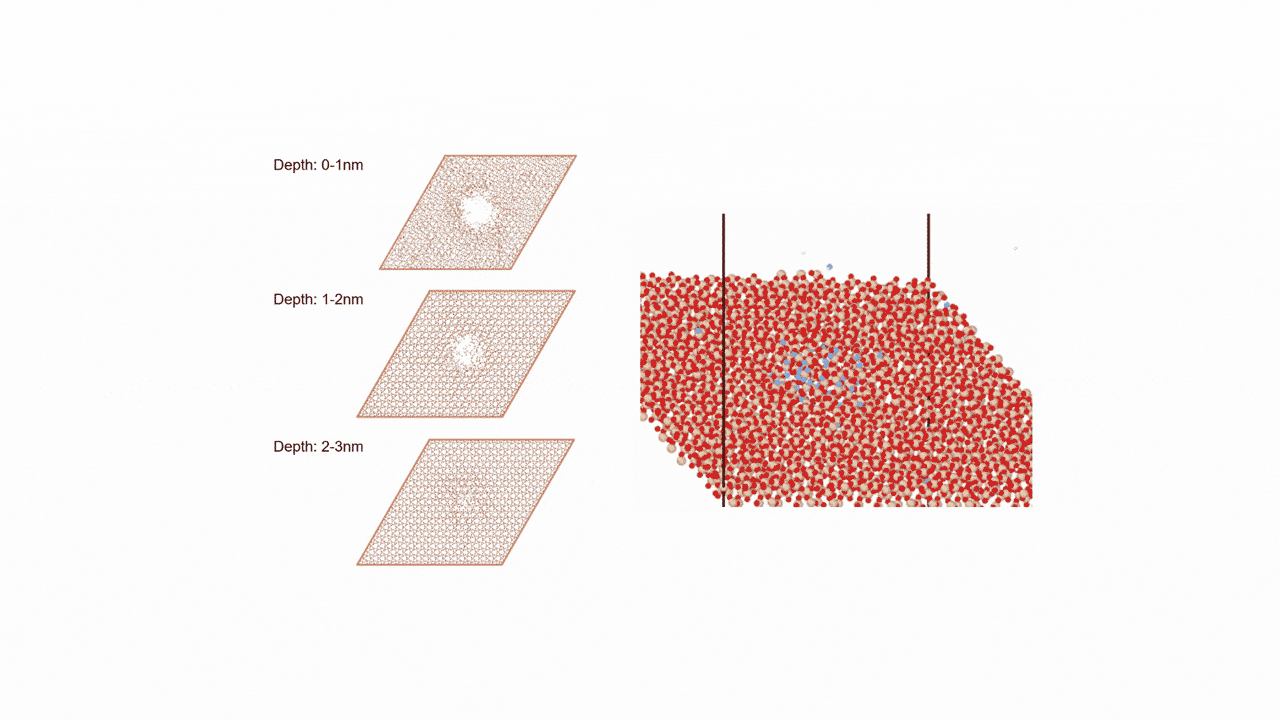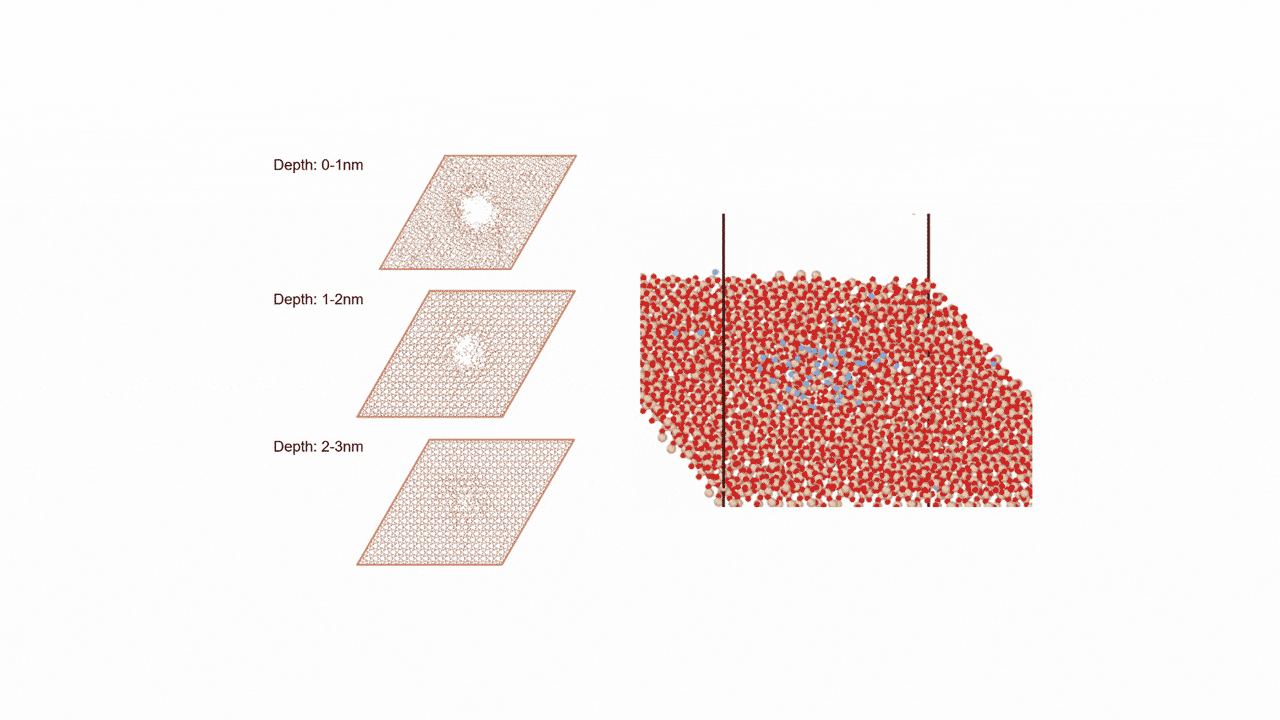
MD計算の結果、CaSi2にFを挿入すると、F密度の高い領域でCaF2の2原子層構造ができ、そこからSiが層間を拡散することで構造相転移することがわかった。
この結果から、結晶性材料の格子間にイオン性原子を挿入するような、電荷バランスの変化を伴う計算について、PFPの計算によって実験と対応する結果が得られることが確認できた。
計算条件

参考文献
[1] R. Yaokawa, Nat. Commun. 7, 10657 (2016). [2] A. Nagoya, Phys. Chem. Chem. Phys., 23, 9315 (2021) [3] icet. https://icet.materialsmodeling.orgタグ
本事例の公開日:2024.03.26
-




 URLを
URLを
コピーしました