Molecular dynamics calculations of magnesium ion conducting oxides
Case study provided by: Nagoya Institute of Technology Nakayama lab.
Introduction
There is an urgent need for next-generation batteries that exceed the performance of existing lithium-ion batteries. Among them, magnesium secondary batteries are attracting attention as storage batteries that use inexpensive and abundant elements and are expected to have a greatly improved capacity. However, the low conductivity of magnesium ions in ceramic materials has been a major barrier to its realization.
Atomistic simulation is a promising method for elucidating the conduction mechanism of magnesium ions in ceramics and for searching for new materials. However while conventional first-principles molecular dynamics calculations are highly accurate, they require a long calculation time. Magnesium ions, in particular, tend to interact more strongly with the lattice and have a larger hopping energy. Due to the low site hopping frequency of the ions, the calculations had to be performed at high simulation temperatures, far from the operating temperature, to obtain a statistically significant number of hopping events.
In this study, we used the fast and accurate Matlantis to evaluate the conductivity of magnesium ions using a relatively large structural model.
Calculation Models and Methods
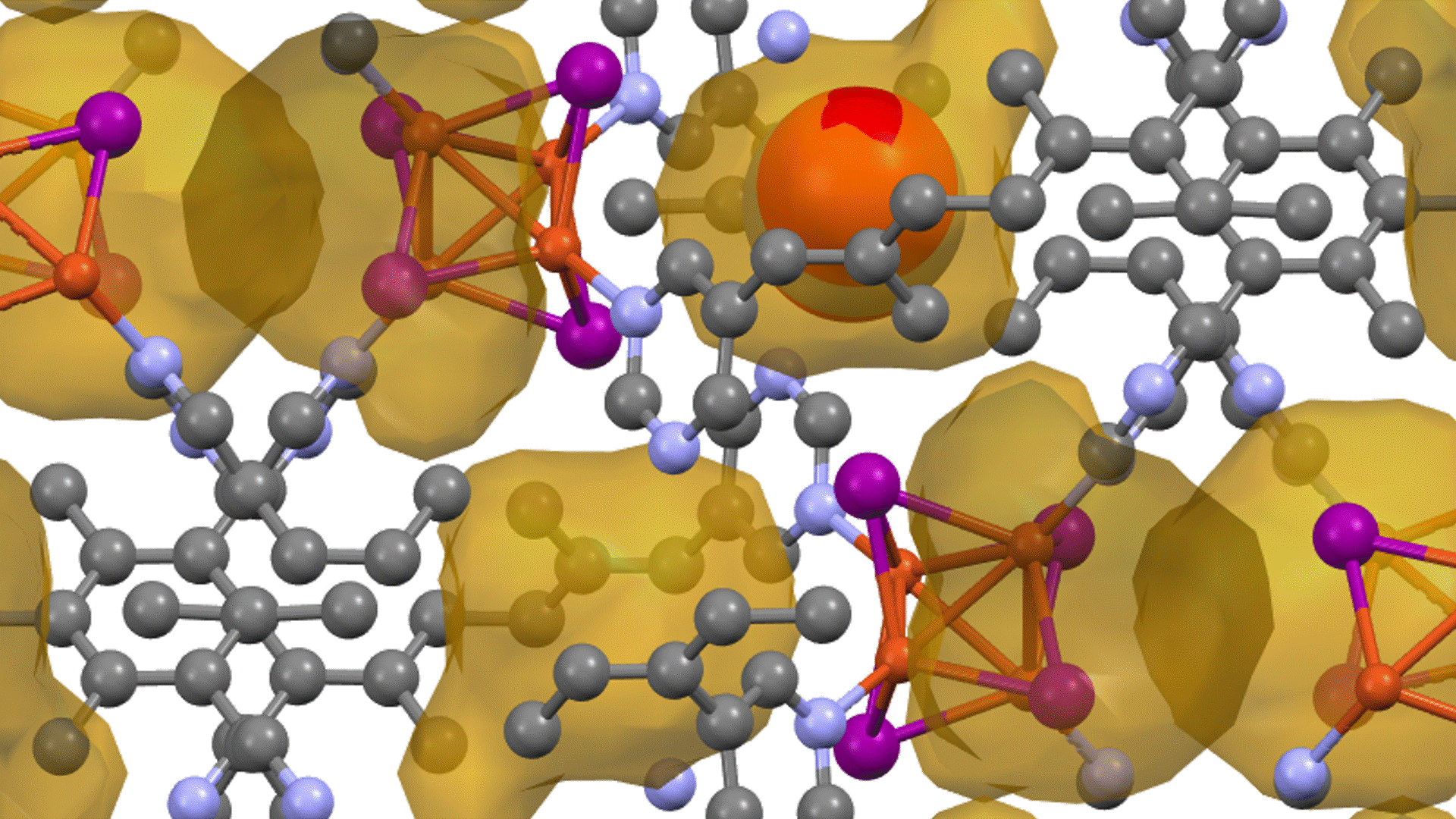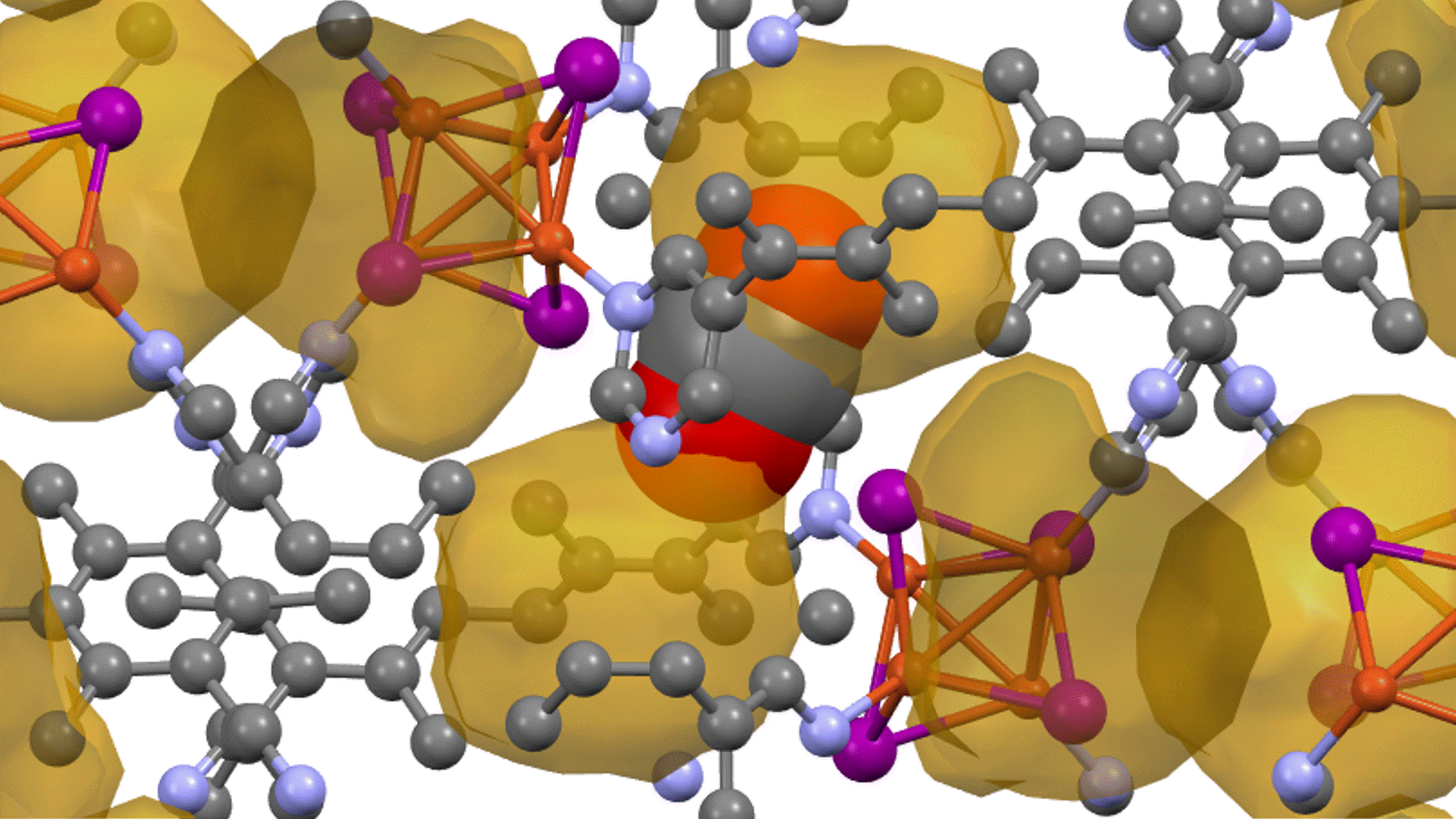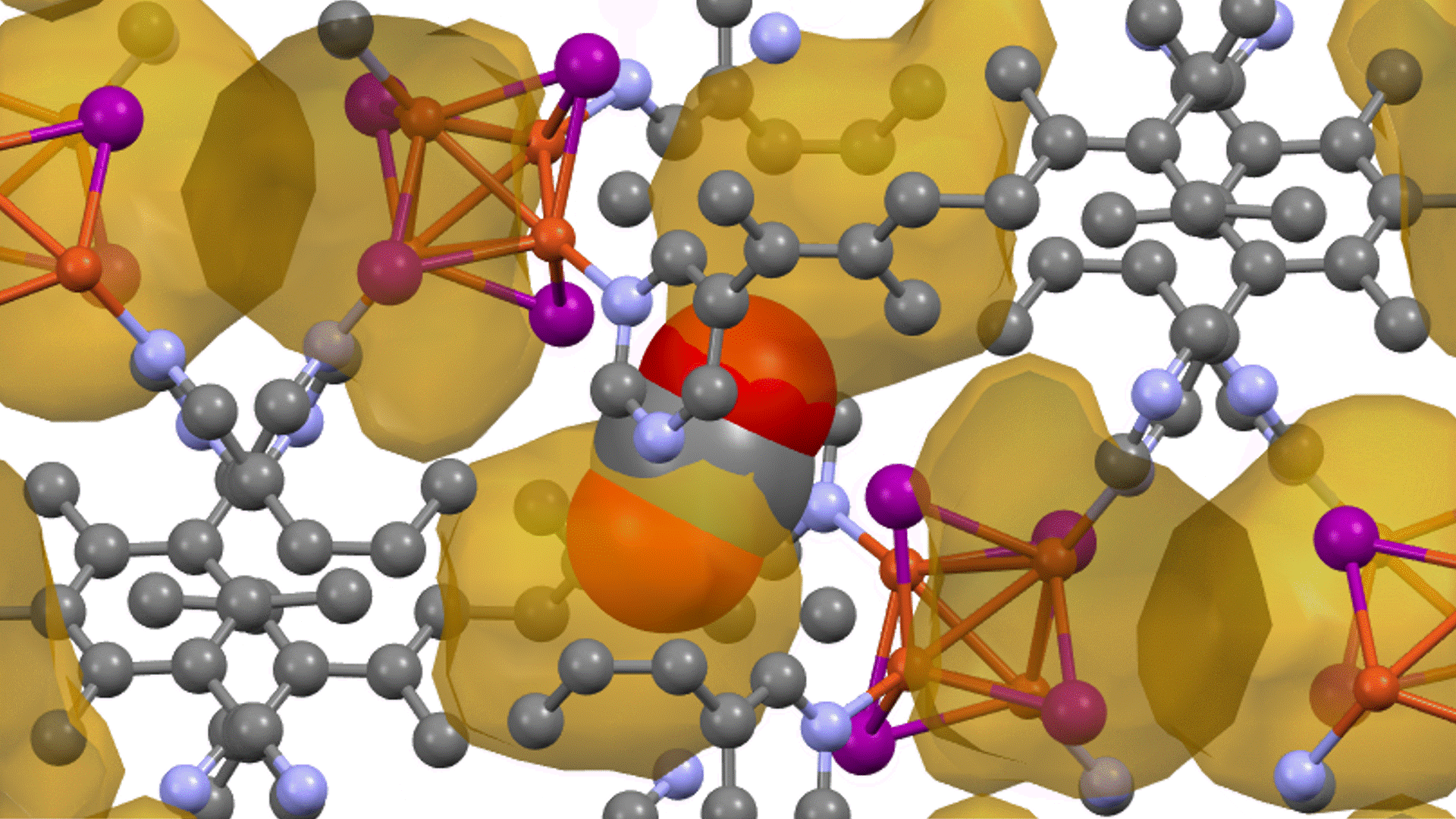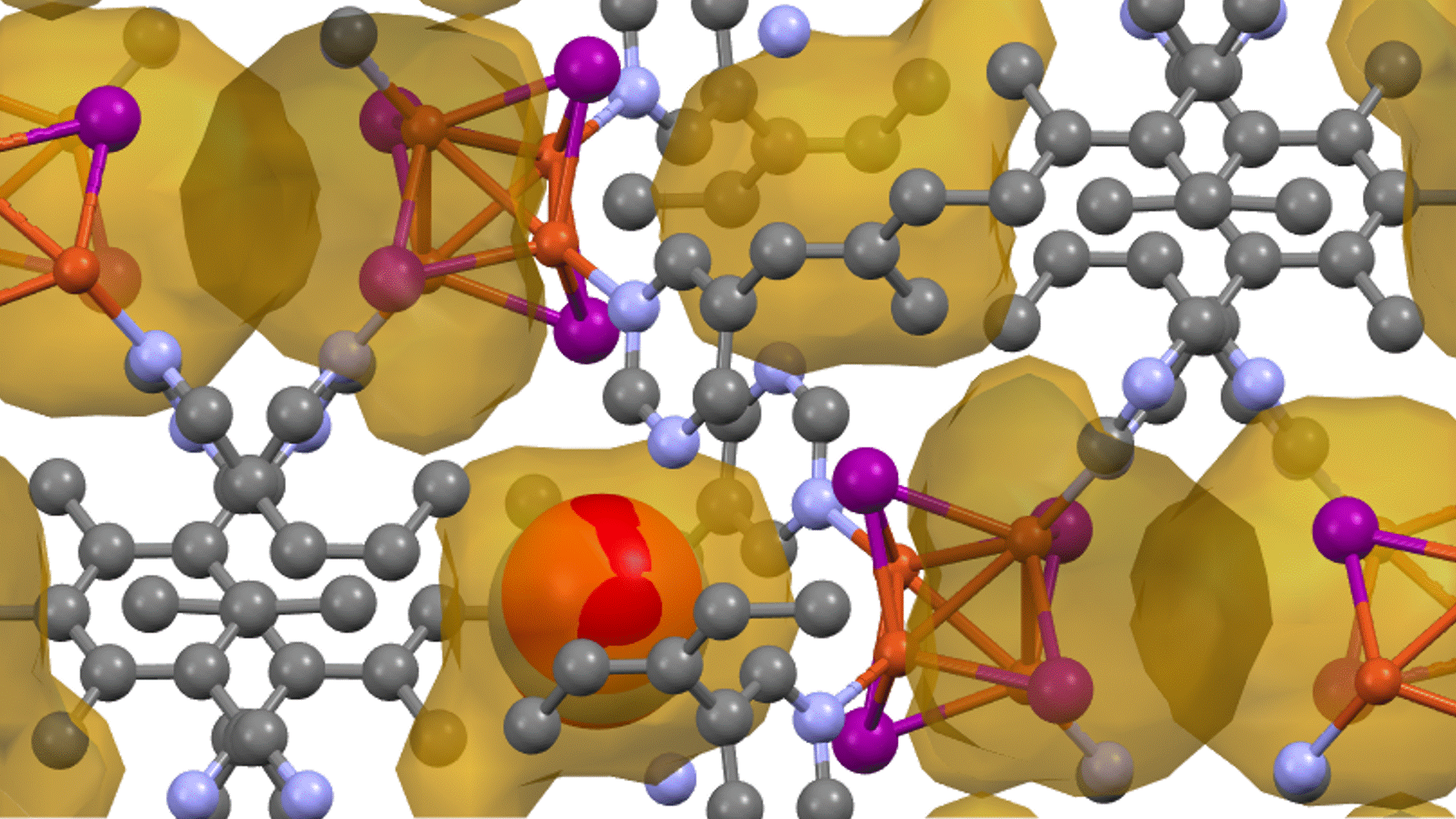
Molecular dynamics (MD) simulations using Matlantis were performed on the ceramic material MgZr4(PO4)6[1,2], which has relatively high Mg ion conductivity. The superstructure model is applied to construct a simulation cell of Mg64Zr384P256O1536, which is a 2240 atom model, and both atomic structure and lattice shape is relaxed to determine the appropriate lattice volume. MD calculations were performed with NVT ensemble (constant volume) at 1073 K.

Results and Discussion
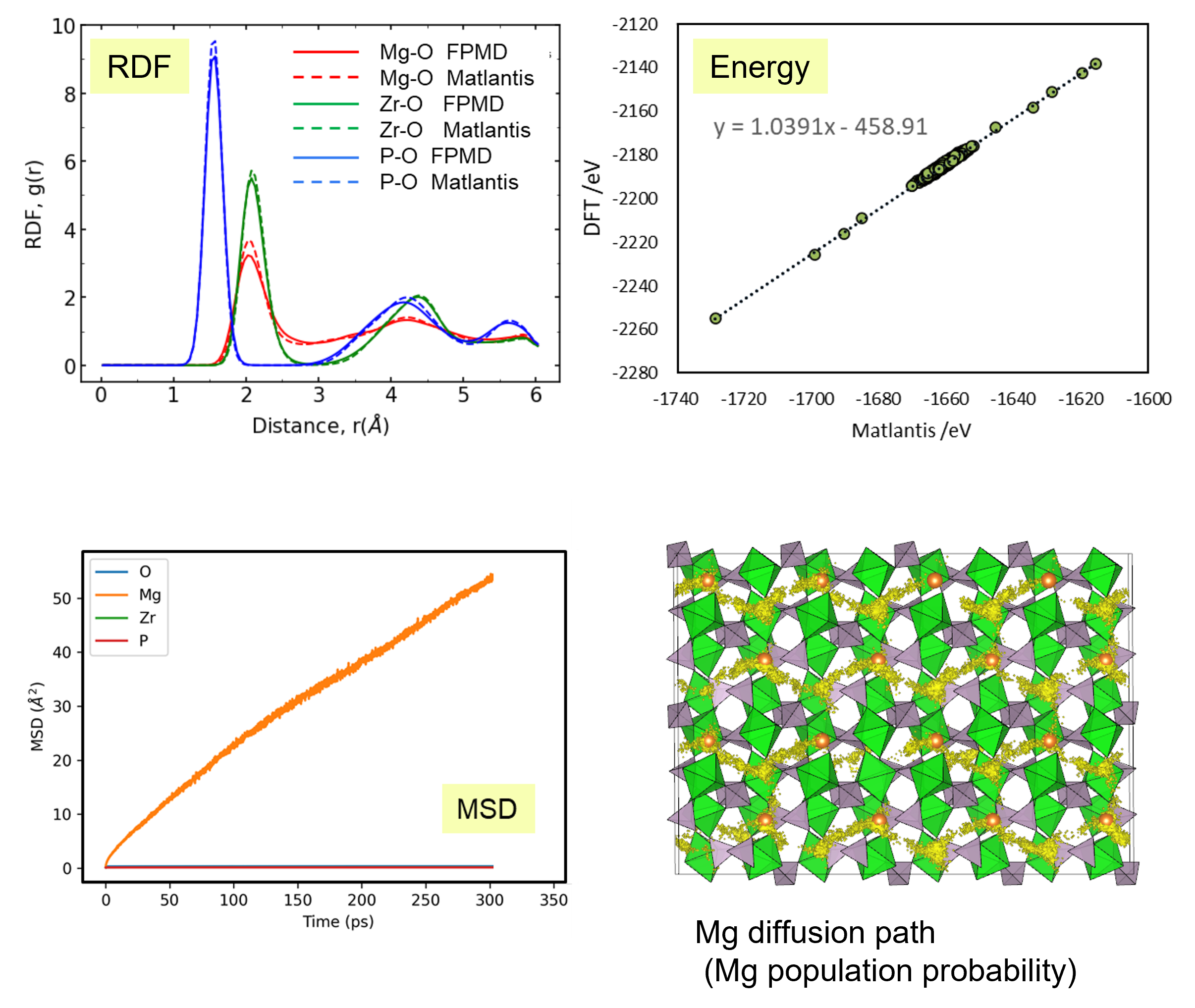
First, we compared the results of Matlantis-based MD with ab-initio MD using a supercell with 280 atoms. Specifically, we evaluated the radial distribution function (RDF), which reflects the bond length distribution between neighbor atoms, and the energy value of the snapshot structure in the MD trajectory generated using Matlantis. The very good agreement was found between Matlantis and ab-intio calculations, confirming that Matlantis achieves the same high accuracy as first-principles calculations.
Based on the above, the diffusion coefficient / diffusion behavior of Mg ions was evaluated by Matlantis. The molecular dynamics simulation was performed at 1073 K using a supercell that contains 2240 atoms. The diffusion of Mg ions was clearly observed from the MSD profile, and the diffusion coefficient could be evaluated with sufficient statistical accuracy.
The supercell used for ab-inito MD contains 280 atoms (Mg8Zr32P48O192), which is relatively large for the quantum mechanical calculations. However, it contains only 8 Mg atoms, which force us to observe the atom hopping and evaluate diffusion coefficient at elevated temperatures 1573~1973 K[2].On the other hand, Matlantis features allow us to use a much larger supercell and much longer simulation time, which enable us to perform simulations at a temperature closer to the operating temperature and obtain statistically accurate results.
References
[1] N. Imanaka, Y. Okazaki and G. Adachi, “Divalent Magnesium Ionic Conduction in Mg1-2x(Zr1-xNbx)4P6O24 (x = 0-0.4) Solid Solutions”, Electrochem. Solid-State Lett., 3, 327-329 (2000) https://iopscience.iop.org/article/10.1149/1.1391138 [2] K. Nakano, Y. Noda, N. Tanibata, M. Nakayama, K. Kajihara, K. Kanamura, Computational investigation of the Mg-ion conductivity and phase stability of MgZr4(PO4)6u, RSC adv., 9, 12590 (2019) https://pubs.rsc.org/en/content/articlelanding/2019/ra/c9ra00513gProfile of the case study provider
Nagoya Institute of Technology Nakayama Laboratory

tag
Publication date of this case: 2022.03.22
-




 URL
URL
Copied
what's new
NEW
A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
Batteries

SiO₂ Dry Etching Simulation Using LightPFP
Batteries
Analysis of Surface Reaction Mechanism between ALD Precursor and Substrate
Batteries
Catalyst Screening for Ammonia Synthesis Catalyst
Batteries
Analysis of CO2 adsorption dynamics of MOF using NEB method
Batteries