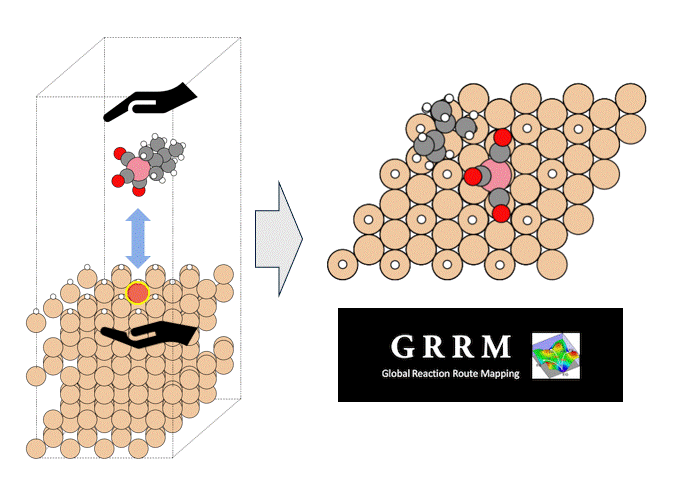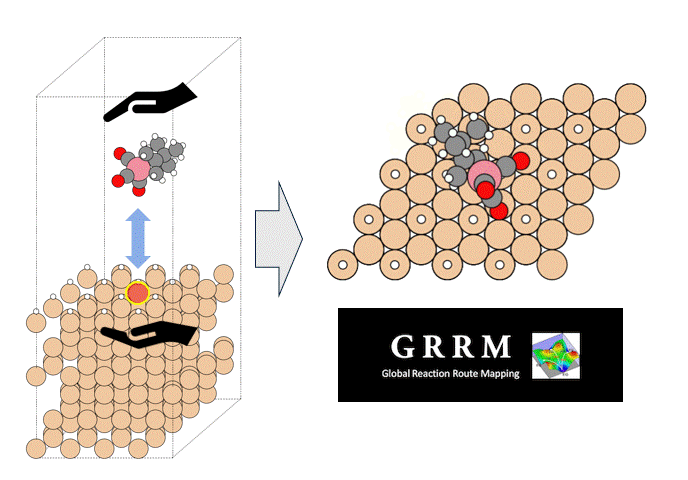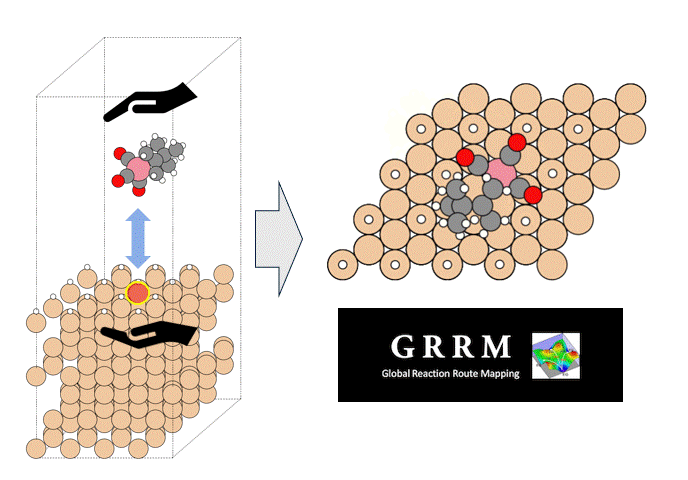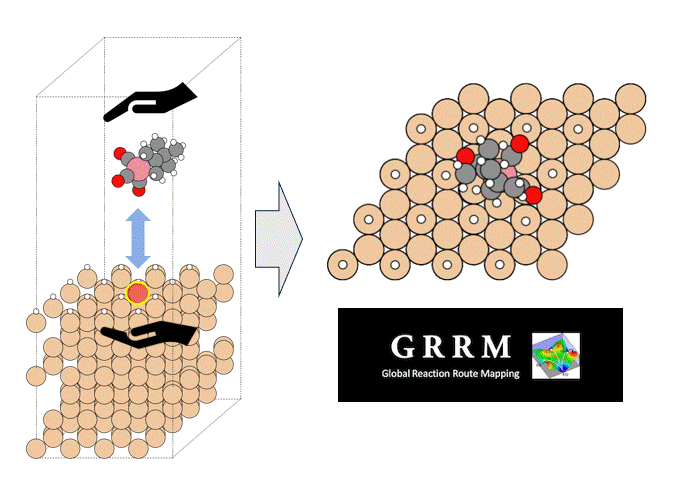
テーマ概要
リチウムイオン電池の後継となる新しい電池の研究開発が注目されています。その中で、安価で豊富な元素を用い、大幅な容量の向上が見込まれる蓄電池としてマグネシウム二次電池が注目されています。しかし、セラミックス材料中でのマグネシウムイオンの伝導性が低く、実現への大きな壁となっています。
材料シミュレーションは、セラミックス中でのマグネシウムイオンの伝導機構の解明や新材料探索に期待される手法ですが、従来の第一原理分子動力学計算は高精度である反面、計算時間が長いという問題を抱えていました。特にマグネシウムイオンは、格子との相互作用が強く、ホッピングエネルギーが大きくなる傾向にあります。そのため、イオンのサイトホッピング頻度が低いことから、統計的に有意なホッピングのイベント数を得るために、使用温度からかけ離れた高温で計算をしなければならないなどの制約がありました。
今回、高速かつ高精度なMatlantisを使って、比較的大きな構造モデルを用い、マグネシウムイオンの伝導性を評価しました。
計算モデルと計算方法
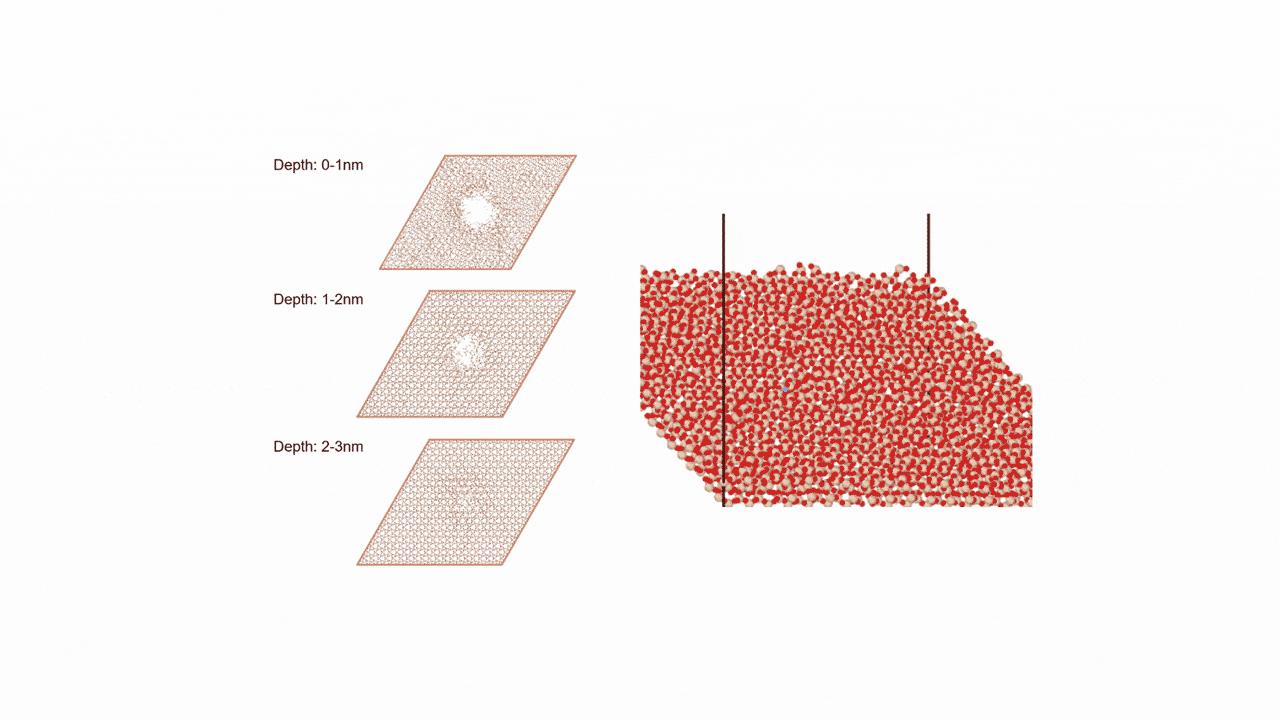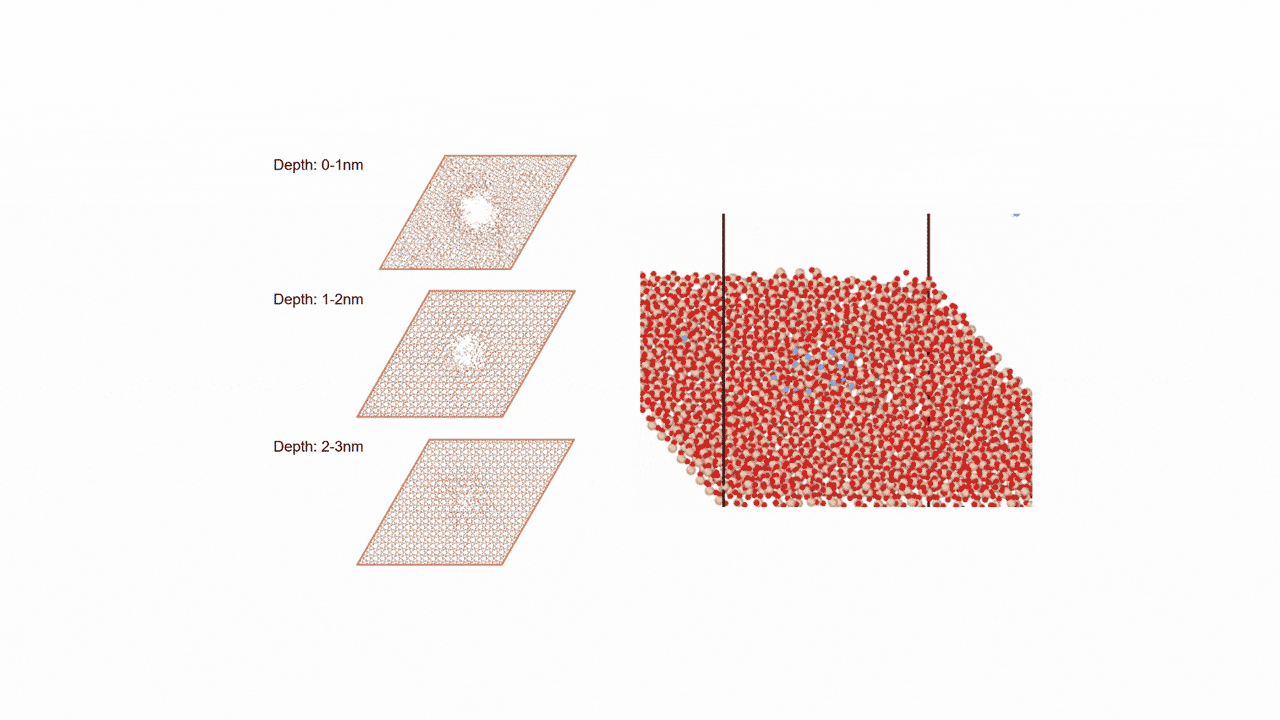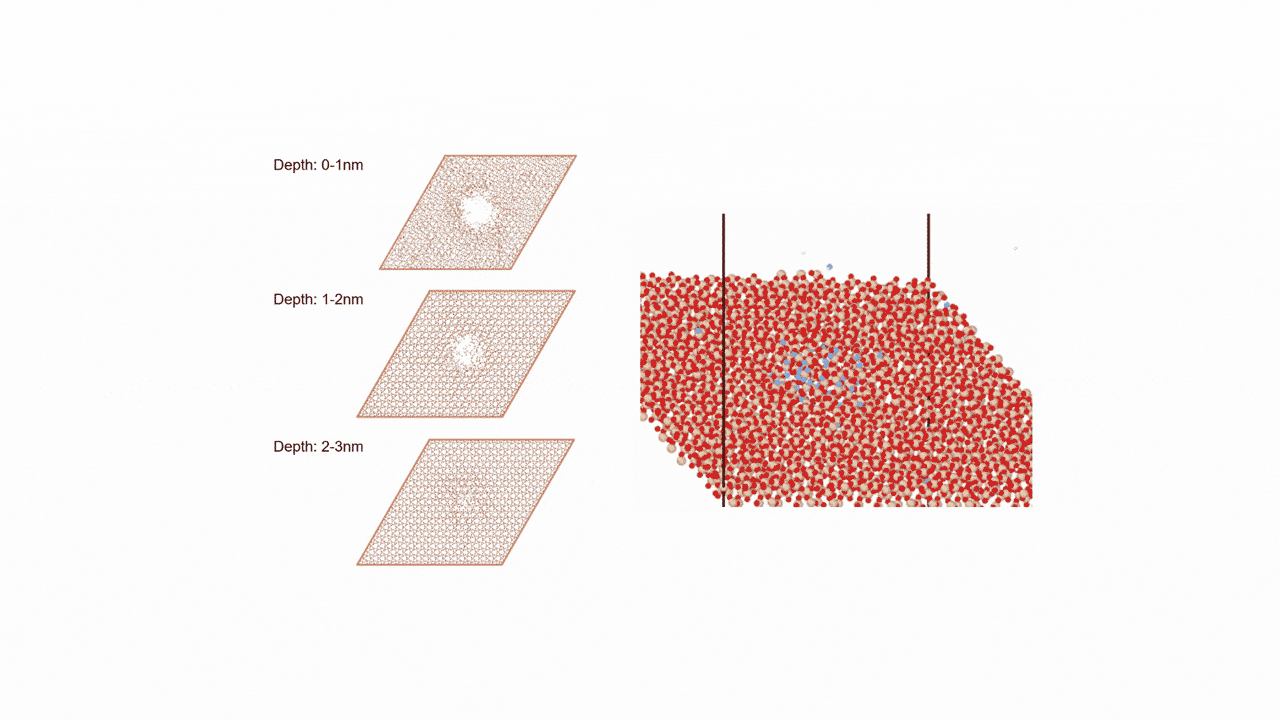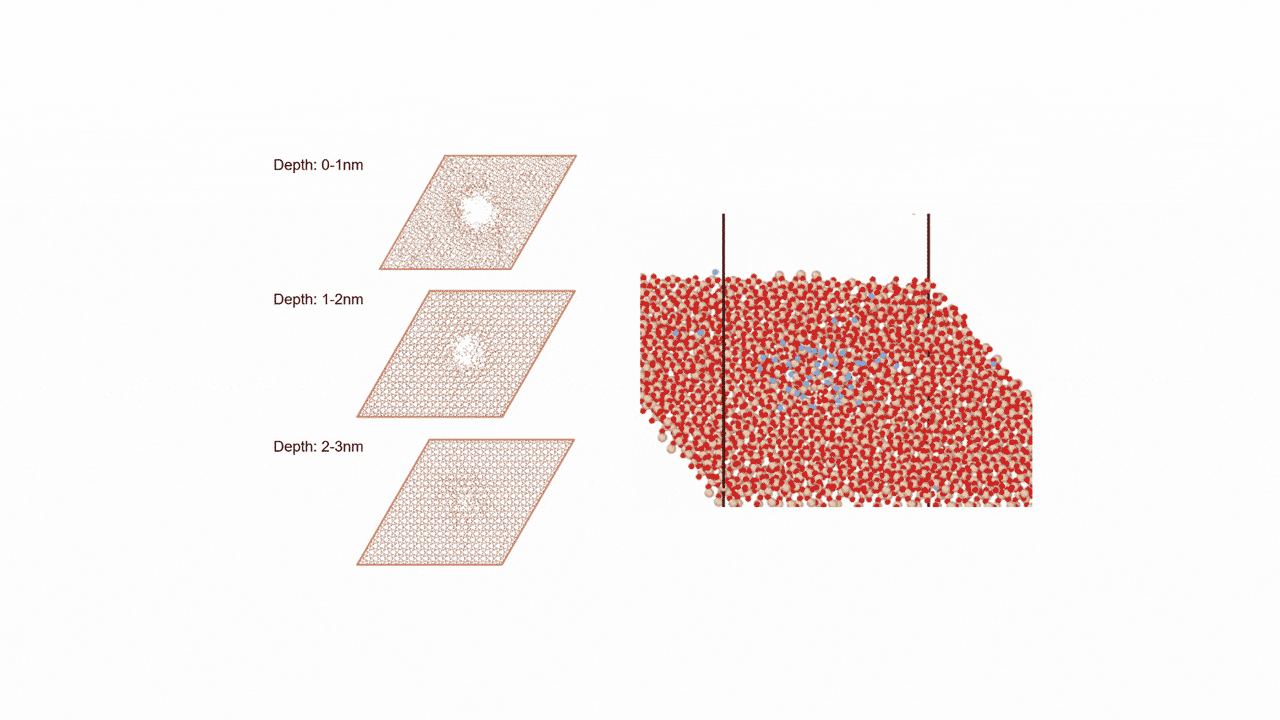
比較的高いMgイオン伝導性を有するセラミックス材料 MgZr4(PO4)6[1,2]に対して、Matlantisを用いた分子動力学(MD)シミュレーションを行いました。超構造モデルを適用し、2240原子モデルとなるMg64Zr384P256O1536のシミュレーションセルを構築し、構造緩和を実行して格子体積を決定します。NVTアンサンブル(体積一定)を適応し、温度は1073 Kの条件でMD計算を実施しました。

計算結果と展望
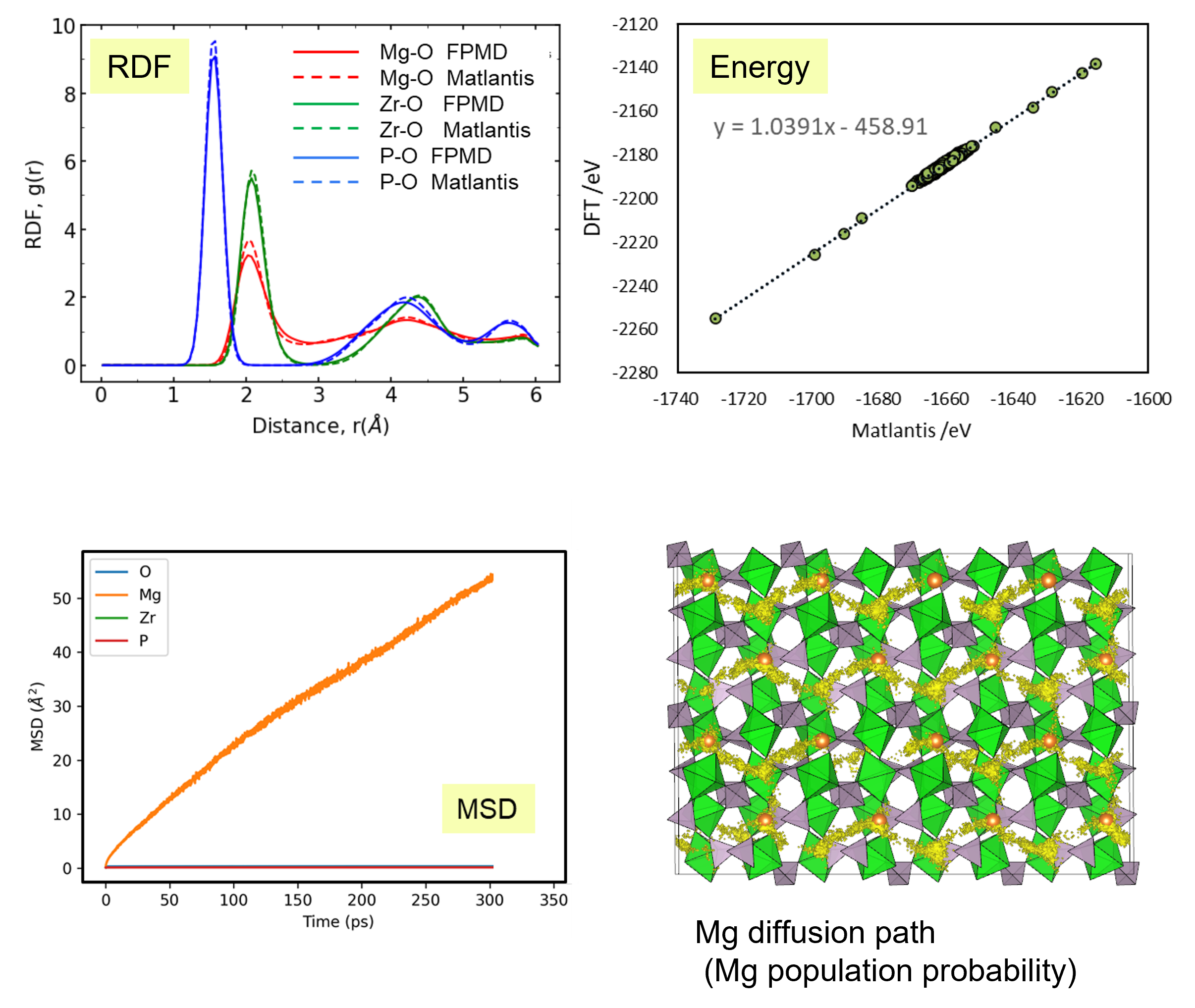
最初に第一原理分子動力学法が適用可能なサイズのセル(280原子)を用いて、MatlantisによるMD計算の結果を比較しました。具体的には、構成するイオン間の結合長分布を反映する動径分布関数(RDF)と、MatlantisによるMDで出力された各時刻におけるスナップショット構造のエネルギー値で評価しました。両者で非常に良い一致が確認され、Matlantisが第一原理計算と同程度の高い精度での計算を実現していることが確認されました。
以上を踏まえて、Mgイオンの拡散評価をMatlantisで実施しました。設定温度を1073 K, 格子サイズを2240として分子動力学法を行ったところ、MSDプロファイルから明瞭なMgイオンの拡散が得られ、十分な統計精度で拡散係数を評価できることが分かりました。
第一原理分子動力学計算ではモデル格子がMg8Zr32P48O192(原子数280ケ)となり量子力学計算をする対象としては比較的大きなモデルでありながらMgはわずかに8ケしか含まれないため、ホッピングイベントを観測するためにシミュレーション温度を1573~1973 Kに設定して評価する必要がありましたが[2]、Matlantisではモデル格子サイズ、シミュレーション時間を十分に大きくできるため、使用温度により近い設定でも十分な統計精度でシミュレーションが可能になりました。
参考文献
[1] N. Imanaka, Y. Okazaki and G. Adachi, “Divalent Magnesium Ionic Conduction in Mg1-2x(Zr1-xNbx)4P6O24 (x = 0-0.4) Solid Solutions”, Electrochem. Solid-State Lett., 3, 327-329 (2000) https://iopscience.iop.org/article/10.1149/1.1391138 [2] K. Nakano, Y. Noda, N. Tanibata, M. Nakayama, K. Kajihara, K. Kanamura, Computational investigation of the Mg-ion conductivity and phase stability of MgZr4(PO4)6u, RSC adv., 9, 12590 (2019) https://pubs.rsc.org/en/content/articlelanding/2019/ra/c9ra00513g事例提供者プロフィール
名古屋工業大学 中山研究室

タグ
本事例の公開日:2022.03.22
-




 URLを
URLを
コピーしました