Surface Reaction Dynamics of ZnDTP Lubricant Additive
Case Study Provider: University of Hyogo・Washizu Lab.
Theme Overview
Lubricants are used in various products such as automobiles, construction machinery, and home appliances. They are indispensable in keeping machinery running smoothly and preventing mechanical failure.
Lubricating oils generally contain additives to improve performance. ZnDTP (zinc dialkyldithiophosphate) is one of the most important additives. Its action has been studied for a long time, and it is recognized that ZnDTP is accompanied by complex material changes through chemical reactions. Molecular simulations have also been applied to elucidate the detailed mechanisms. However, the fact that ZnDTP is composed of multiple elements has made it difficult to create a high-precision force field.
In this study, we calculated the chemical reaction dynamics of ZnDTP using Matlantis, which allows chemical reactions to be observed and supports to arbitrary combinations of elements.
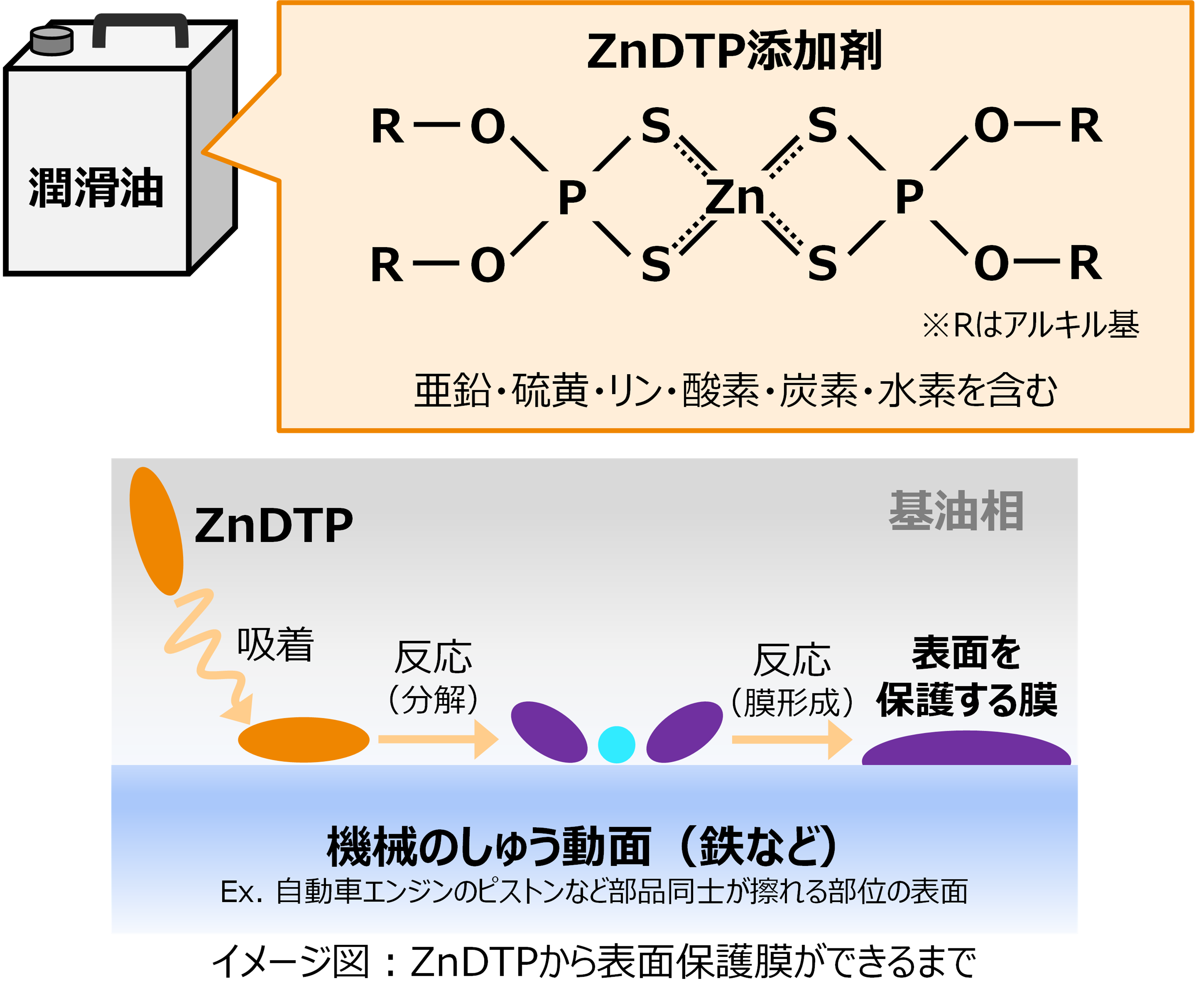
Calculation Models and Methods
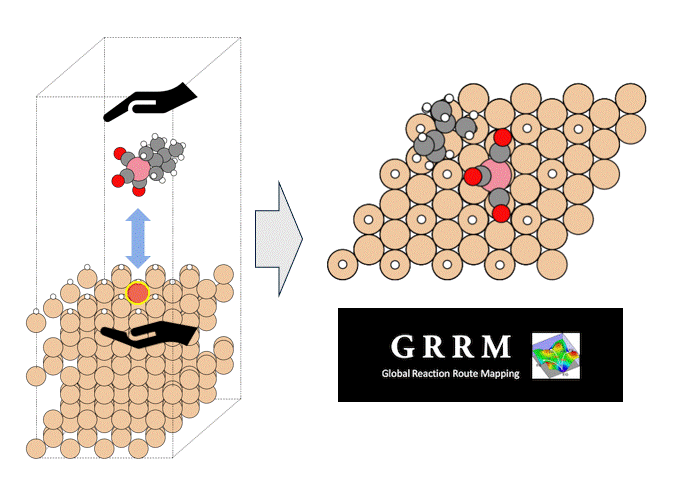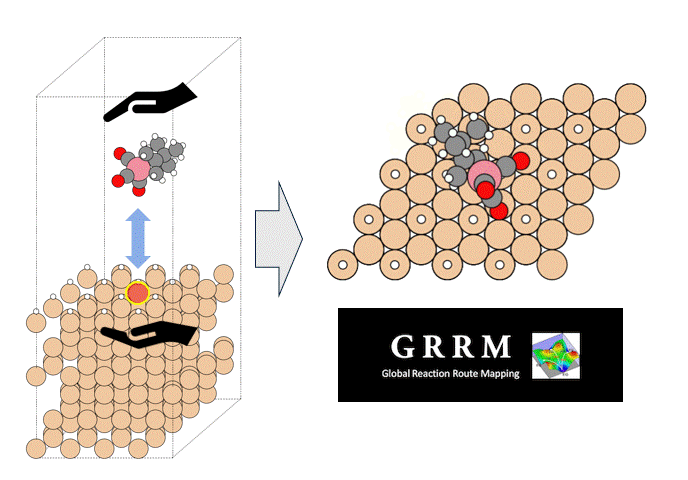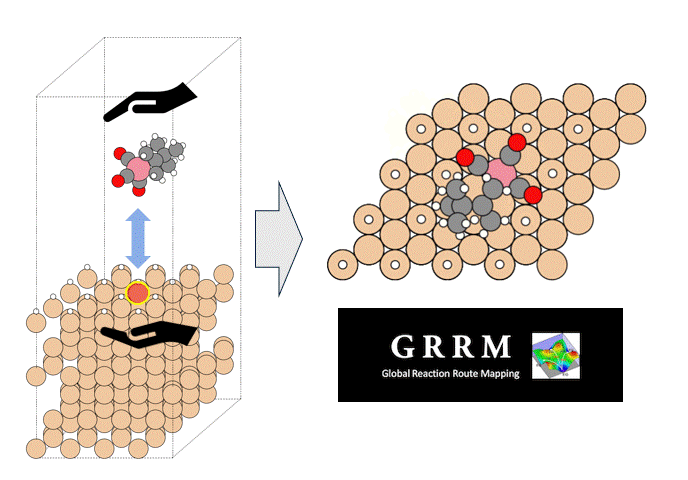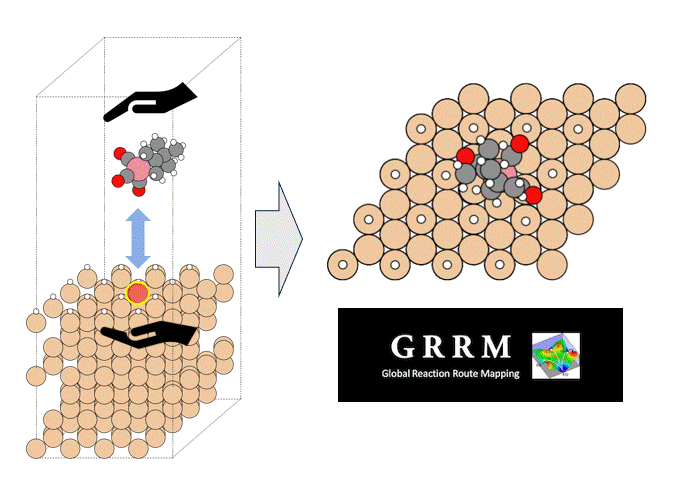
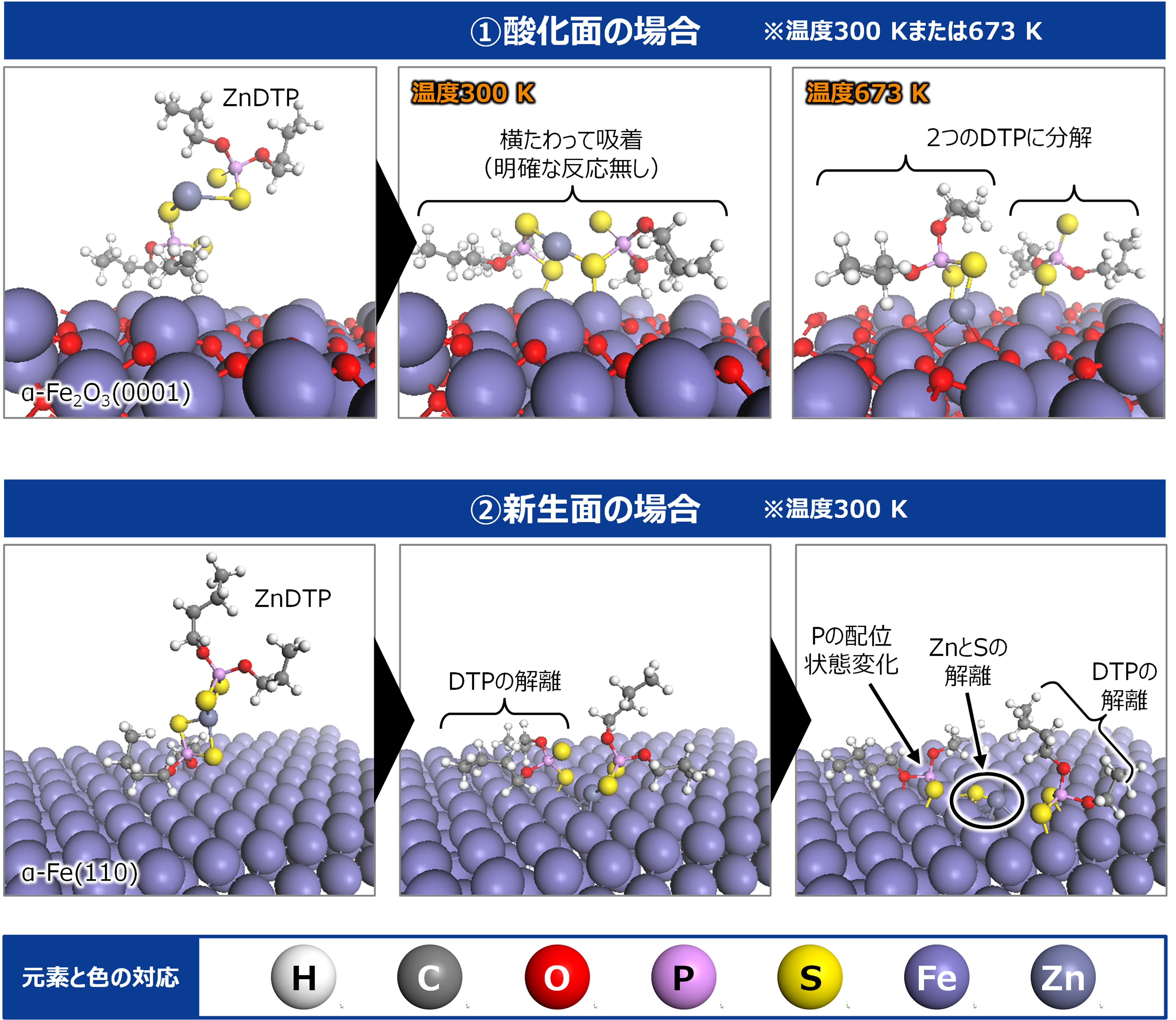
The molecular structure of ZnDTP was created on Matlantis and placed on the α-Fe2O3(0001) surface to simulate the sliding surface of a machine. Four alkyl groups R of ZnDTP are propyl or butyl groups. In addition, a model with ZnDTP placed on the α-Fe(110) surface was also created to simulate a situation where the native oxide layer on the sliding surface has been removed and a nascent surface has been exposed. Molecular dynamics (MD) simulations were performed on these two models using Matlantis.

Results and Discussion
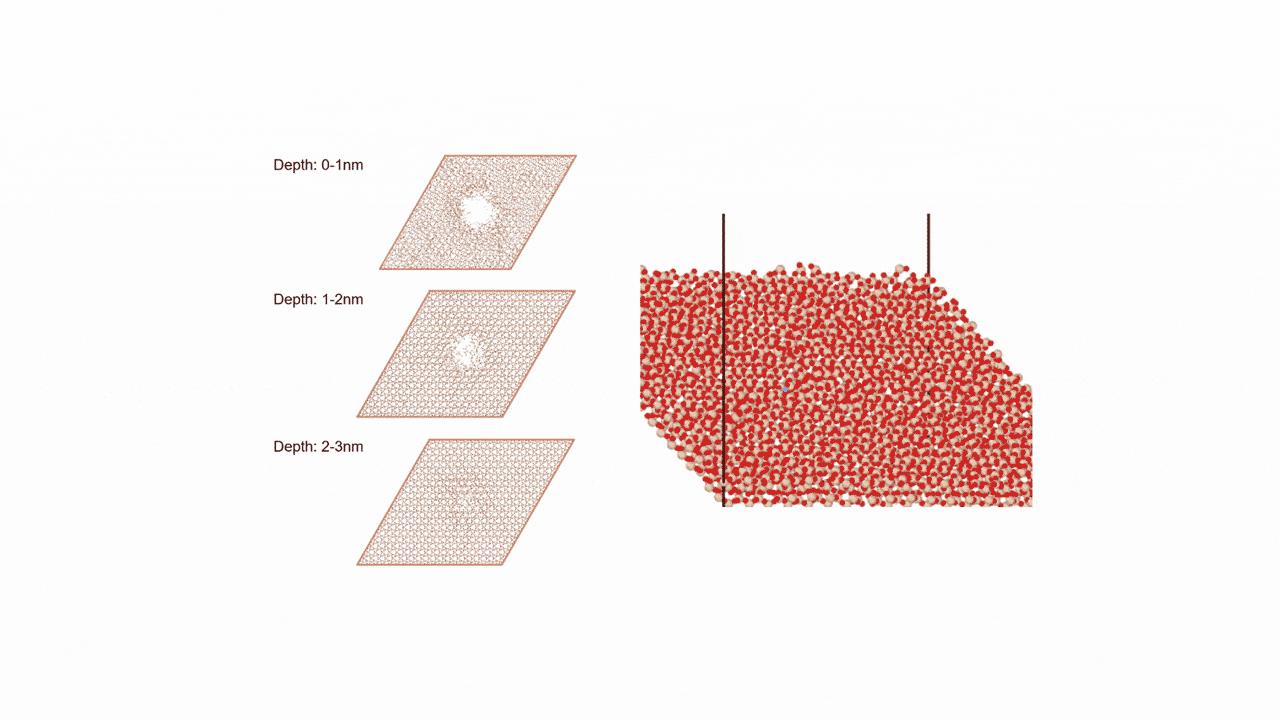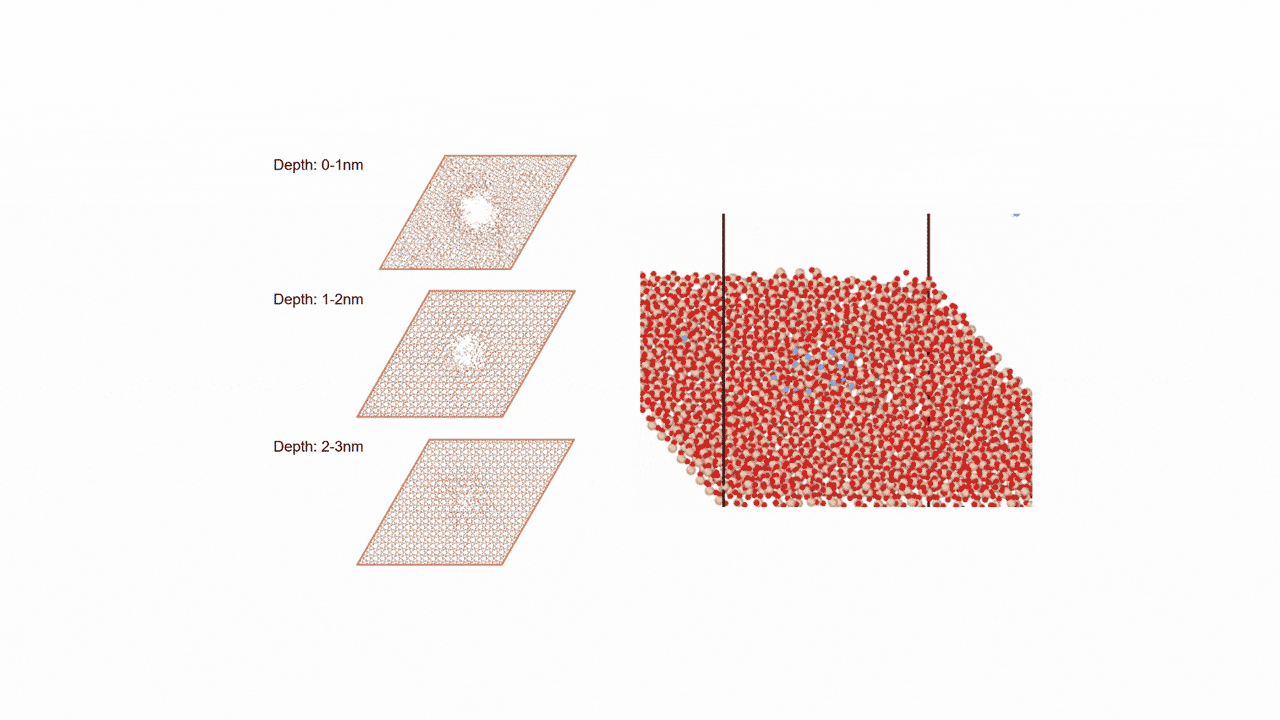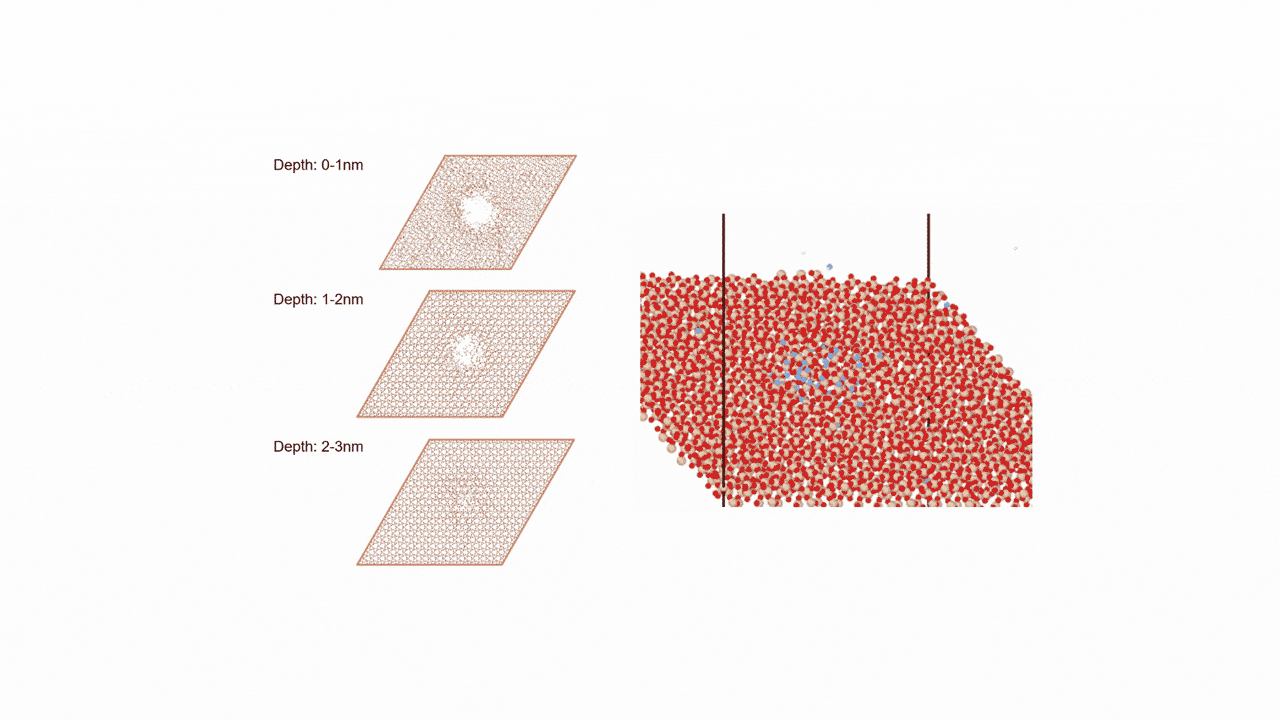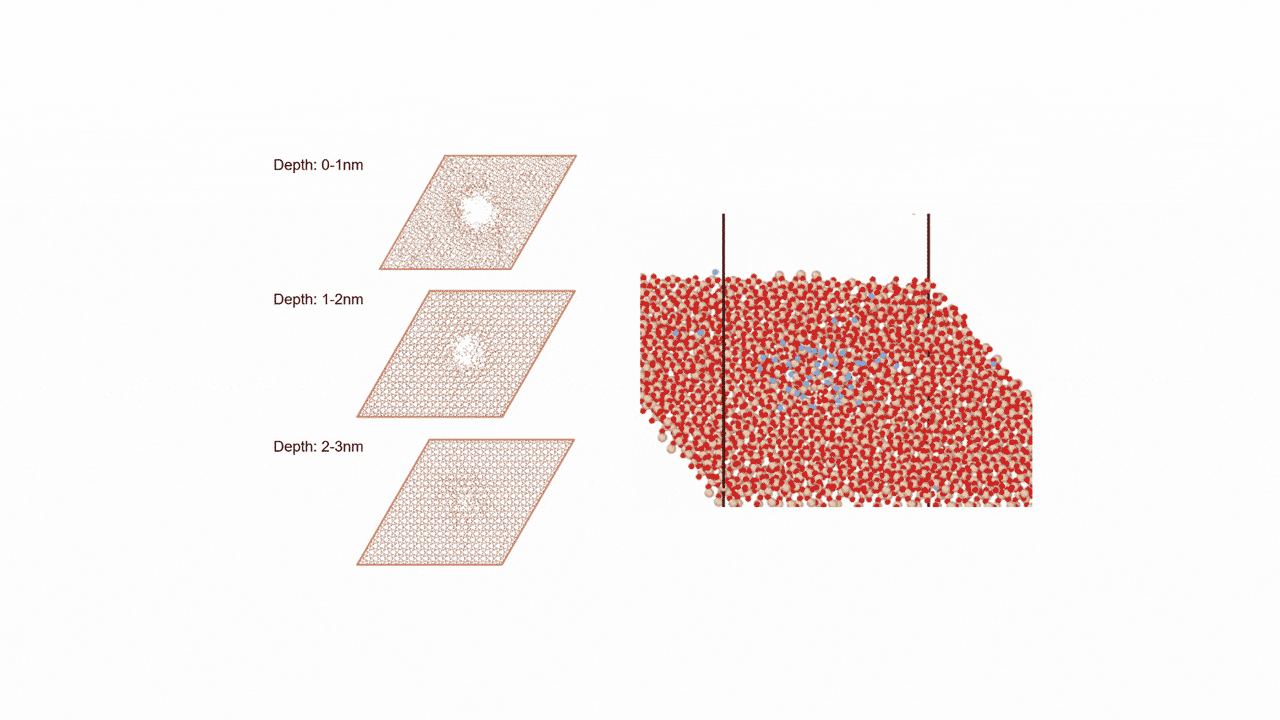
MD simulation results showed that (1) ZnDTP on the α-Fe2O3(0001) surface was only adsorbed and no decomposition reaction was observed. On the other hand, decomposition reactions such as dissociation of DTP and Zn and changes in the coordination state of P were observed on the (2) α-Fe(110) surface with increasing simulation time. Analysis of change in atomic charge revealed that there was almost no charge transfer between ZnDTP and the surface in case (1), whereas a significant charge transfer occurred in case (2). Thus, it was clarified that the surface properties affect the charge state of ZnDTP, which in turn causes differences in reactivity. When the temperature in (1) was set to 673 K, a reaction of decomposition into two DTPs occurred, a result consistent with experimental assumptions [1][2].
As mentioned above, the model handled in this calculation includes seven elements, including zinc, sulfur, phosphorus, oxygen, carbon, and hydrogen, which make up the ZnDTP, plus iron on the surface. Conventional classical MD studies, for example, require the creation of force fields covering multiple element combinations, and a great deal of time is spent before the simulation can begin. Matlantis, on the other hand, does not require the user to create force fields, and thus allows the user to “casually” start simulating chemical reactions in complex systems.
Taking advantage of these strengths, we expect to be able to perform simulations that have been difficult to perform in the past, such as, for example, the synergistic effects of molybdenum-containing additives and ZnDTP, or the clarification of optimal reaction conditions.
References
[1] K. Ito, JM Martin, C. Minfray, K. Kato, “Formation Mechanism of a Low Friction ZDDP Tribofilm on Iron Oxide”, Tribol Trans., 50, 211-216 (2007). https://doi.org/10.1080/10402000701271010 [2] H. Spikes, “The History and Mechanisms of ZDDP”, Tribol. Lett., 17, 469-489 (2004). https://link.springer.com/article/10.1023/B:TRIL.0000044495.26882.b5Profile of the case study provider
University of Hyogo, Washizu Laboratory
tag
Publication date of this case: 2023.03.16
-




 URL
URL
Copied
what's new
NEW
Calculation and Verification of Methanol Synthesis Catalysts
Methanol synthesis catalystsLarge-scale screening
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis

SiO₂ Dry Etching Simulation Using LightPFP
Semiconductor
Analysis of Surface Reaction Mechanism between ALD Precursor and Substrate
SemiconductorsReaction Pathway Exploration