Surface composition analysis of Cu-Indium intermetallic compound under reaction temperature
Waseda University SEKINE group

Overview
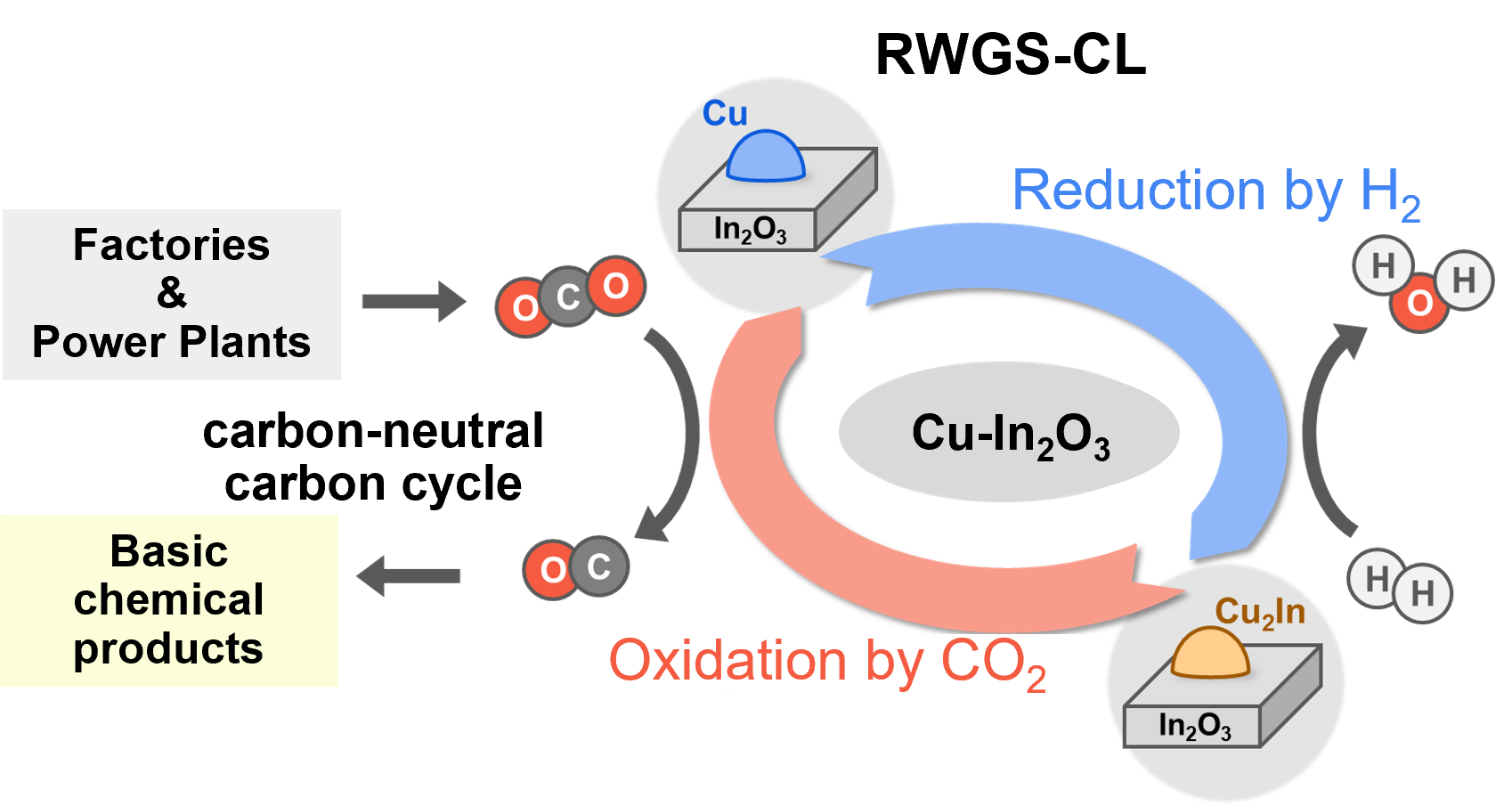
Significant reduction of CO2, a causative agent of global warming, is essential to achieve carbon neutrality [1].
Among these, the chemical loop-based reverse water gas shift (RWGS-CL) reaction, which converts CO2 to the chemical raw material CO, is considered to make a significant contribution to a sustainable carbon cycle [2].
Our laboratory has previously found that composites of Cu-In intermetallic compounds and In2O3 oxides have high performance in the RWGS-CL reaction [3].
In this study, we performed molecular dynamics (MD) calculations using Matlantis on a nm-order cluster model to clarify the surface state of the Cu-In intermetallic compound at the reaction temperature.
Calculation models and methods
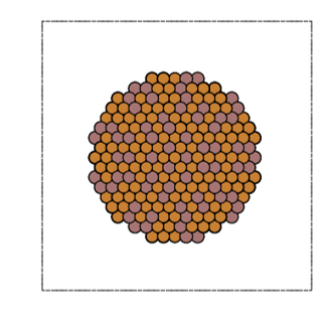
Spherical clusters (radius 19 Å, Cu1311In672) consisting of approximately 2000 atoms were created for an intermetallic compound with a composition of Cu2In, which is expected to exist in the RWGS-CL reaction atmosphere.
While the actual supported metals on catalysts exist in such nm-order clusters, conventional density functional theory (DFT) calculations are computationally expensive for models with such a large number of atoms, and MD calculations are very difficult to perform. However, Matlantis, a high-performance atomistic simulator, still makes such calculations possible.
In this study, we performed MD calculations using the Nosé-Hoover heat bath in Matlantis to evaluate the surface conditions of the intermetallic compound cluster model at 773 K, which is the actual reaction temperature.
Calculation Condition

Calculation Results and Discussion
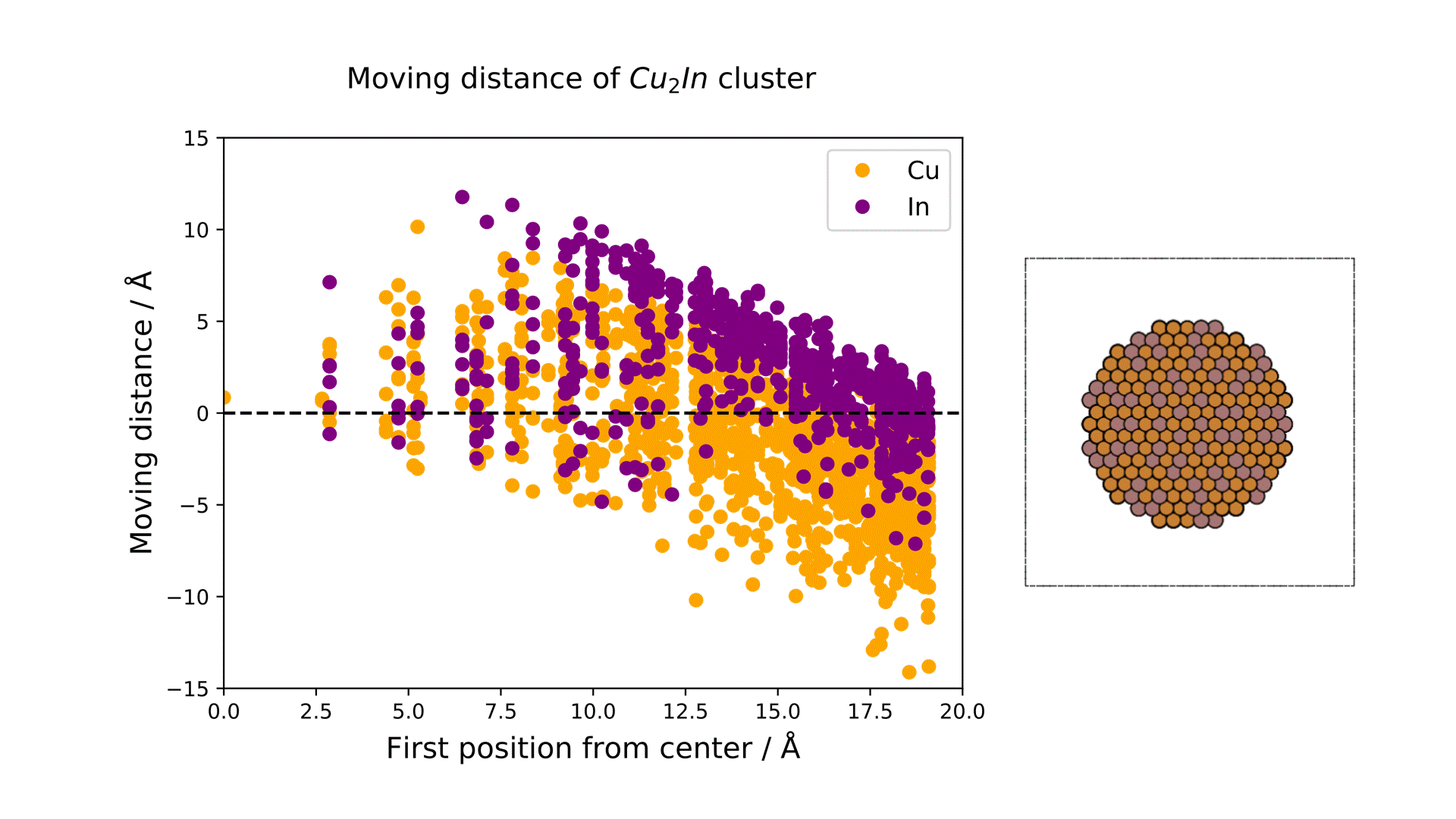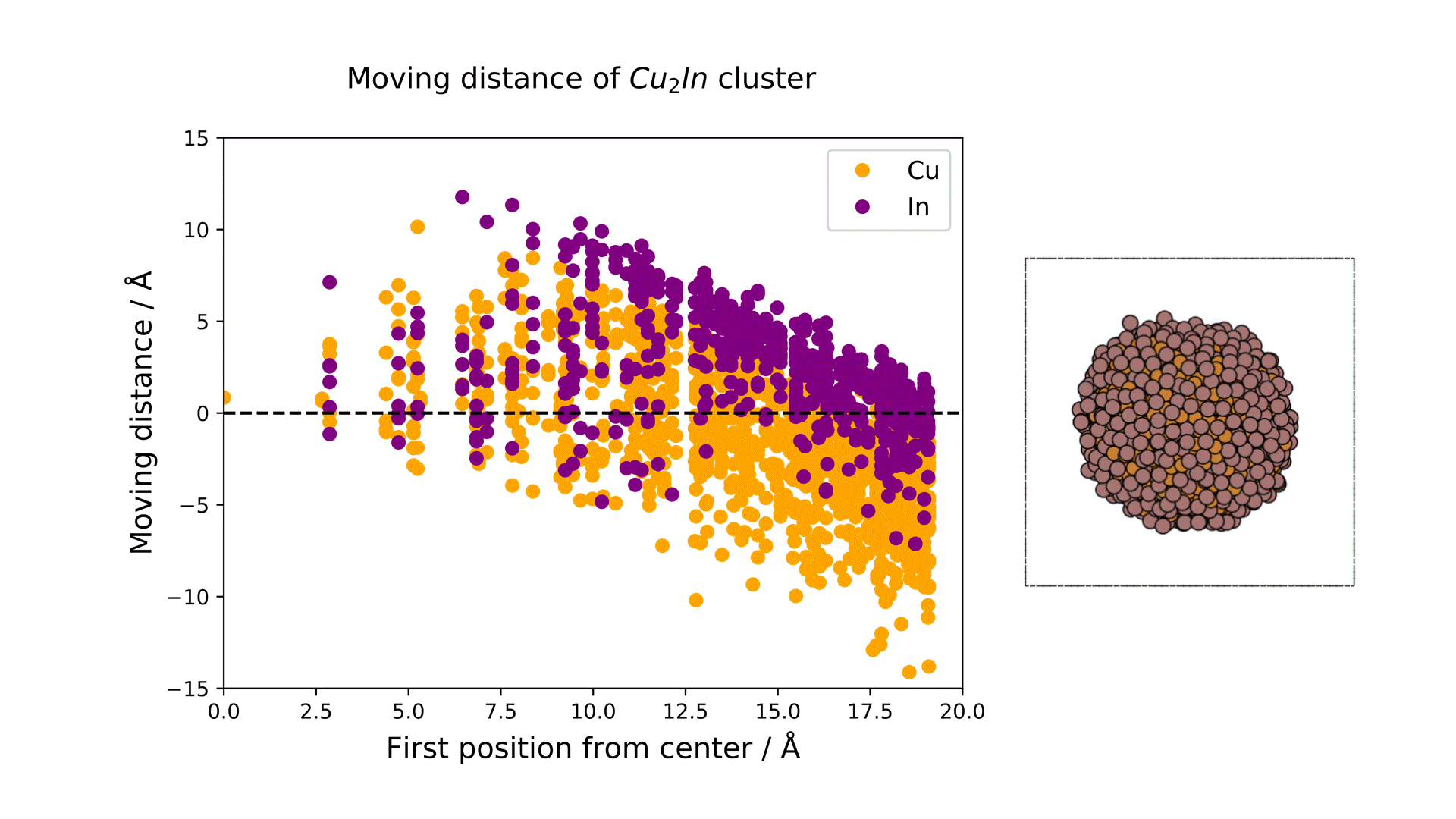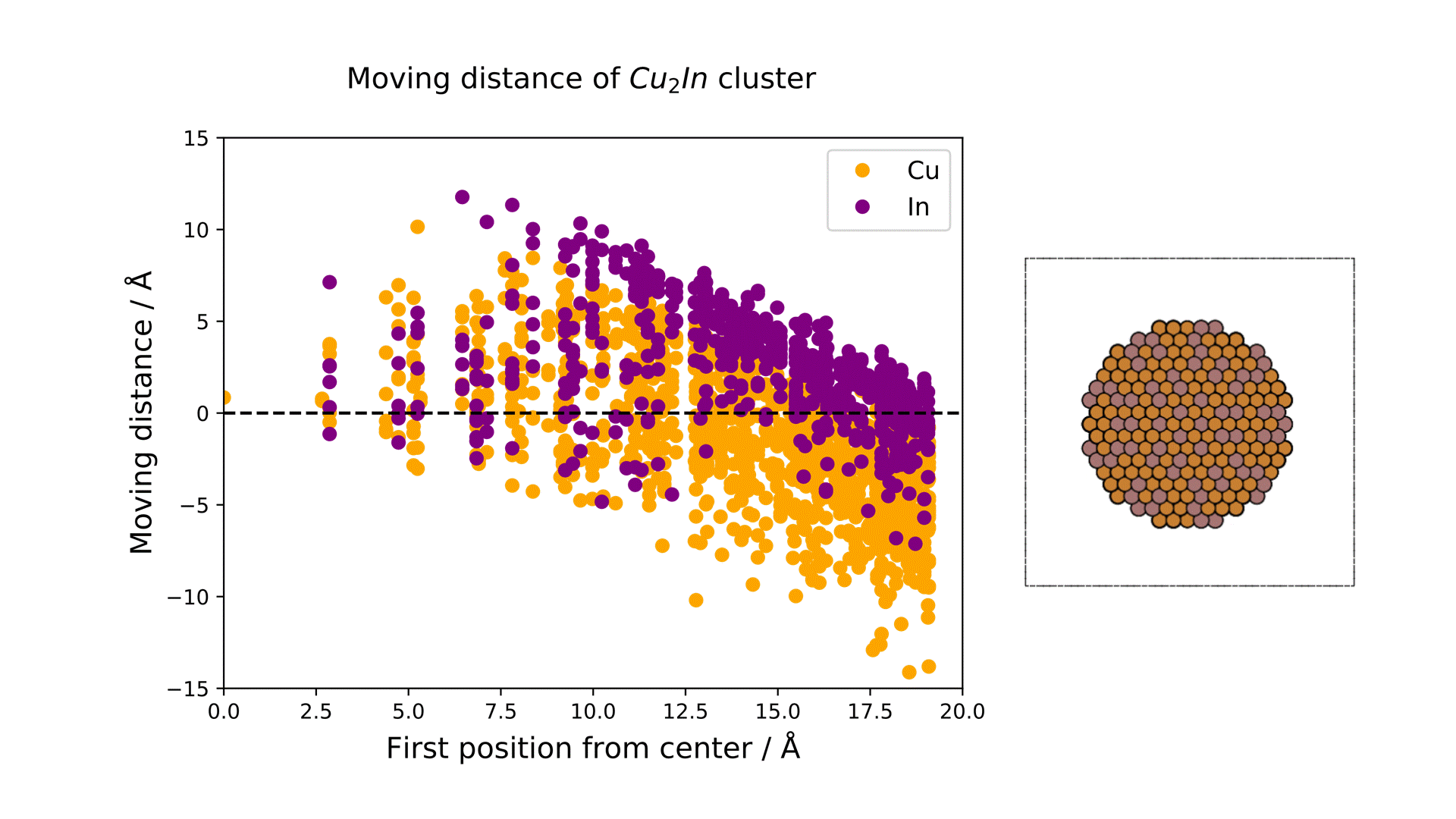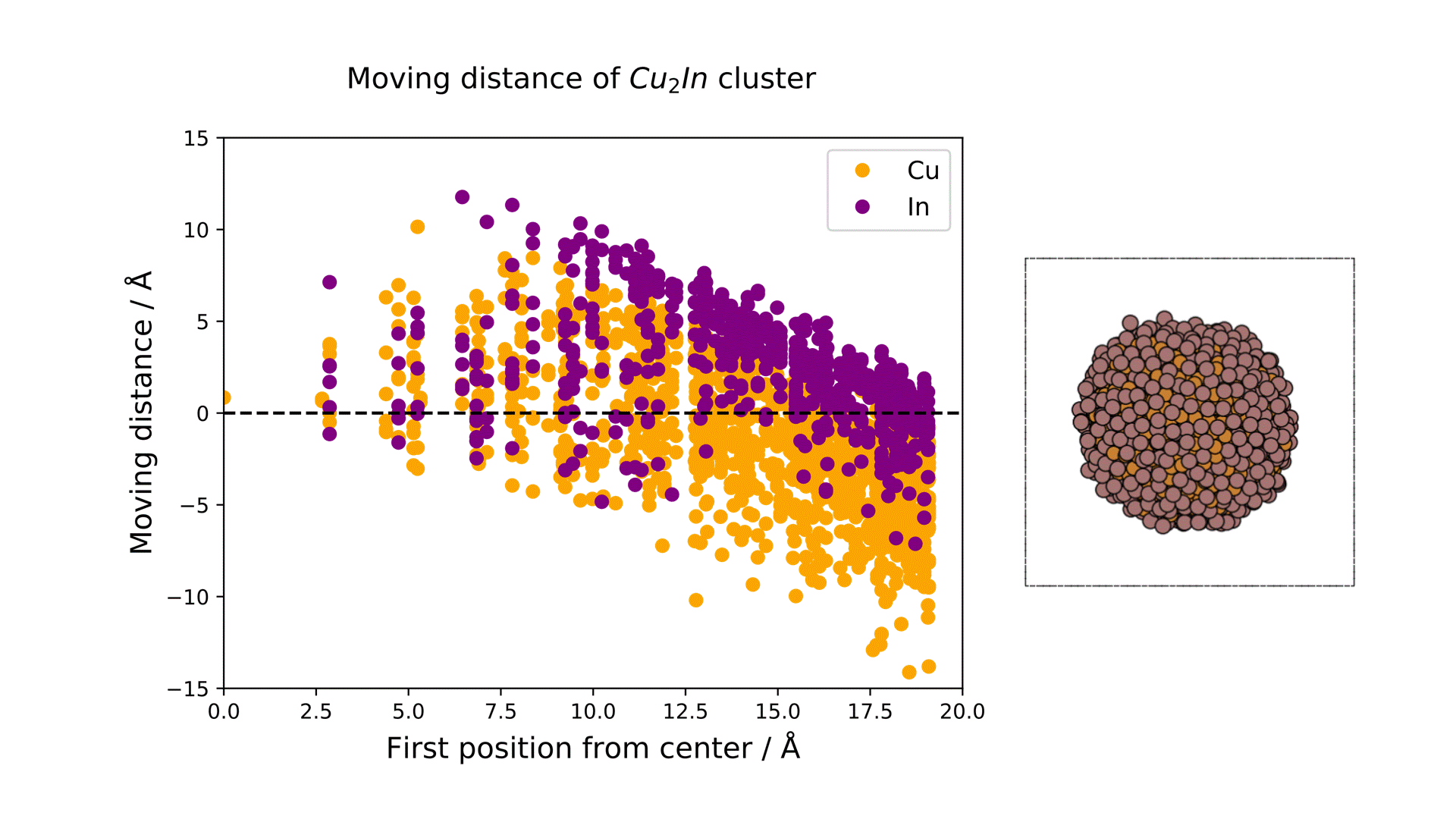
The displacement of each element before and after the MD calculation is shown on the right figure. The horizontal axis shows the distance of each atom from the center of the cluster before the calculation, and the vertical axis shows the radial displacement of each atom in the cluster before and after the reaction.
The results show that both Cu and In exhibit similar displacement behavior near the center of the cluster, while near the surface, In moves outward and Cu moves inward.
This suggests that the Cu-In intermetallic compound at real temperatures has an In-rich alloy structure on the surface, and Matlantis can be used to evaluate the surface structure of models with realistic cluster sizes at real reaction temperatures at low computational cost.
Because Matlantis can calculate models with a greater number of atoms than conventional DFT calculations, we also expect to put it into application to simulate atomic positions and reactivity under experimental conditions by constructing realistic models, such as intermetallic compounds on In2O3.
In addition, by taking advantage of the generalizability of Matlantis, it may be possible to predict materials superior to those previously reported from theoretical calculations by comprehensively examining different compositions.