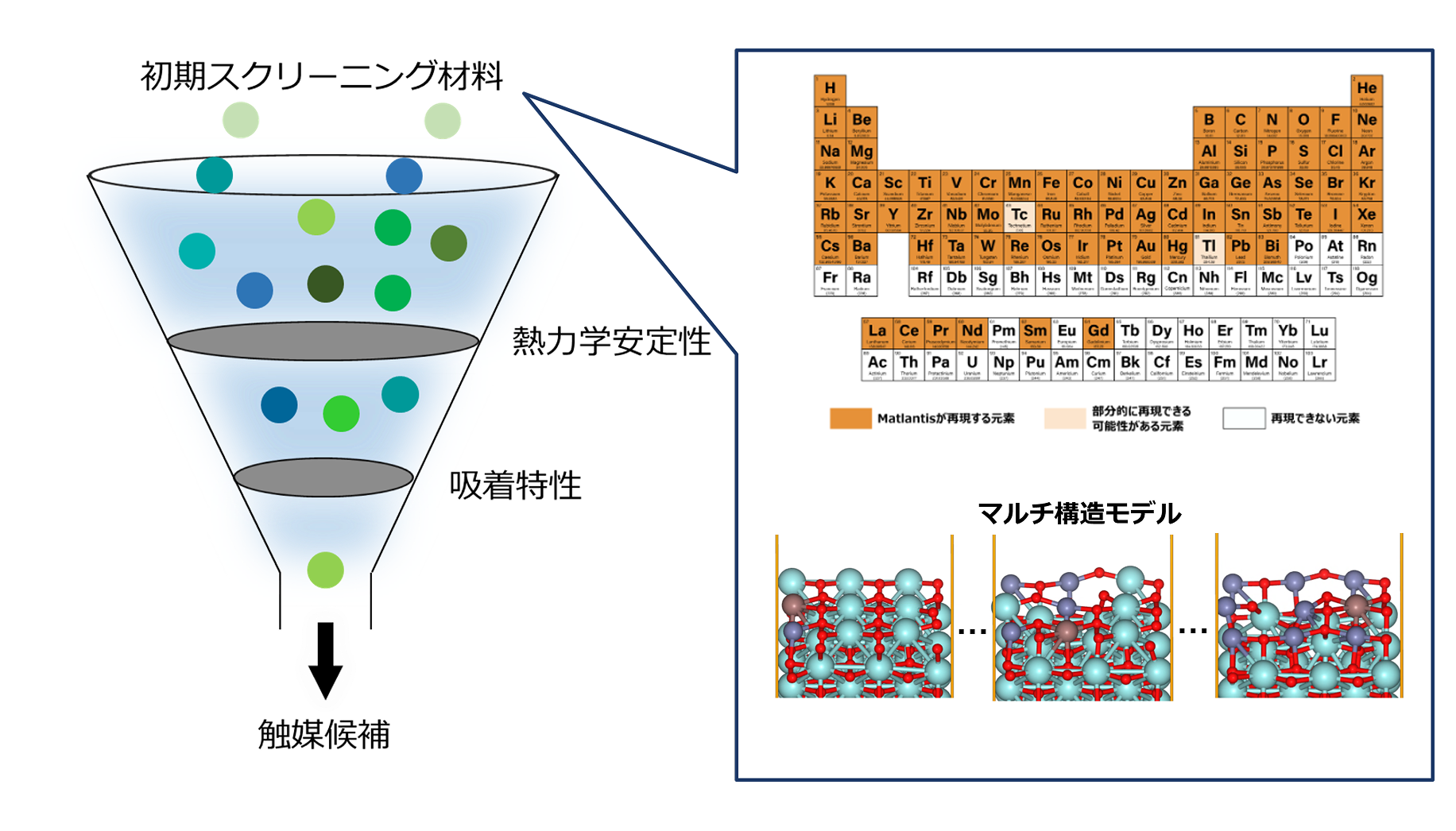
テーマ概要
地球温暖化の原因物質であるCO2を大幅に削減することはカーボンニュートラルの実現のために欠かせません[1]。
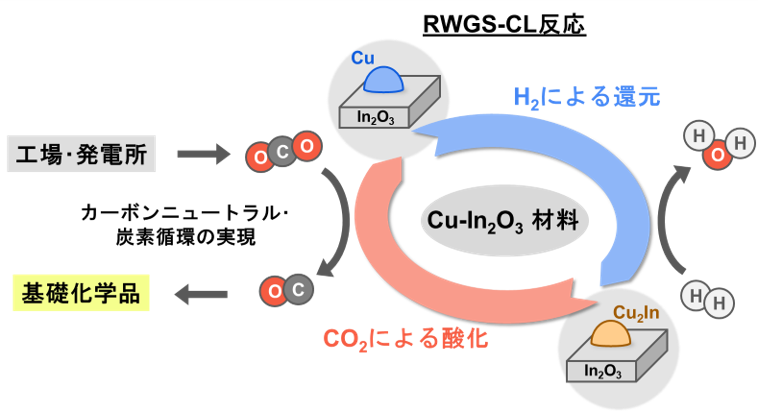
中でもCO2を化学原料であるCOに転換する、ケミカルループを用いた逆水性ガスシフト(RWGS-CL)反応は持続可能な炭素循環に大きく貢献できると考えられています[2]。
当研究室ではこれまでにCu-In金属間化合物とIn2O3酸化物の複合材料がRWGS-CL反応において高い性能を持つ事を見いだしました[3]。
本検討では、このCu-In金属間化合物の反応温度での表面状態を明らかにするために、nmオーダーのクラスターモデルに対してMatlantisを用いて分子動力学(MD)計算を行いました。
計算モデルと計算方法
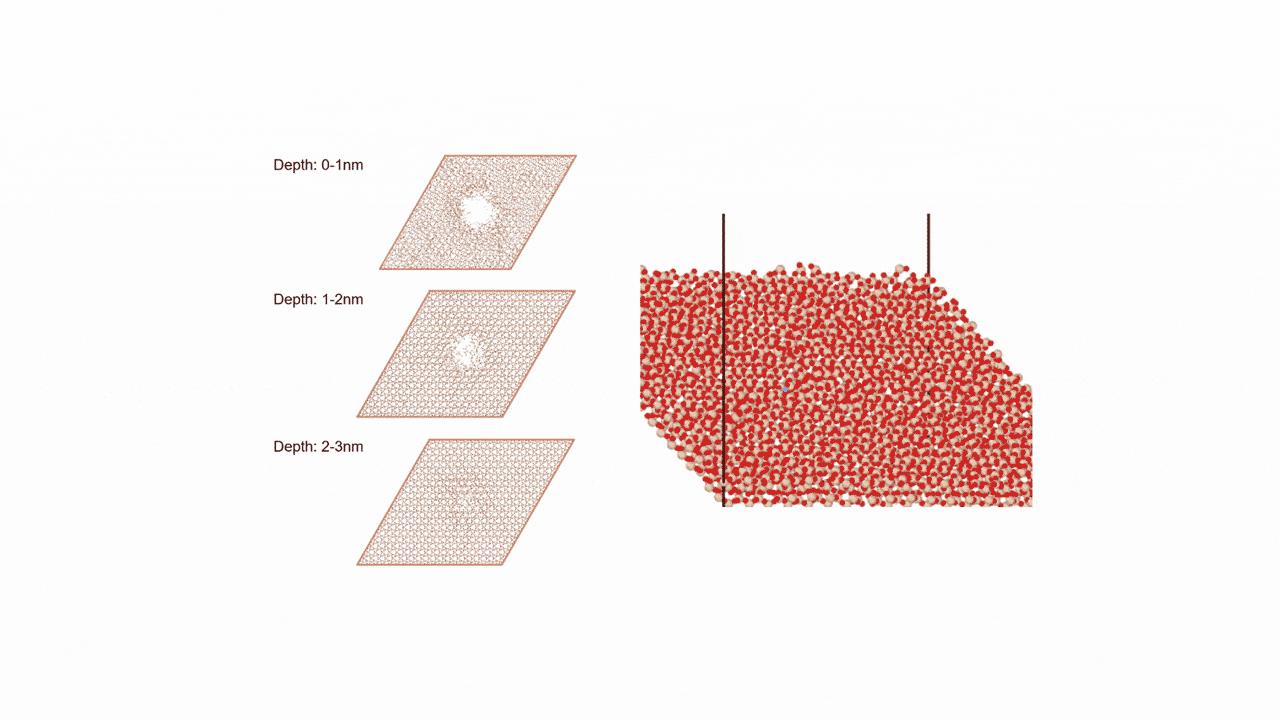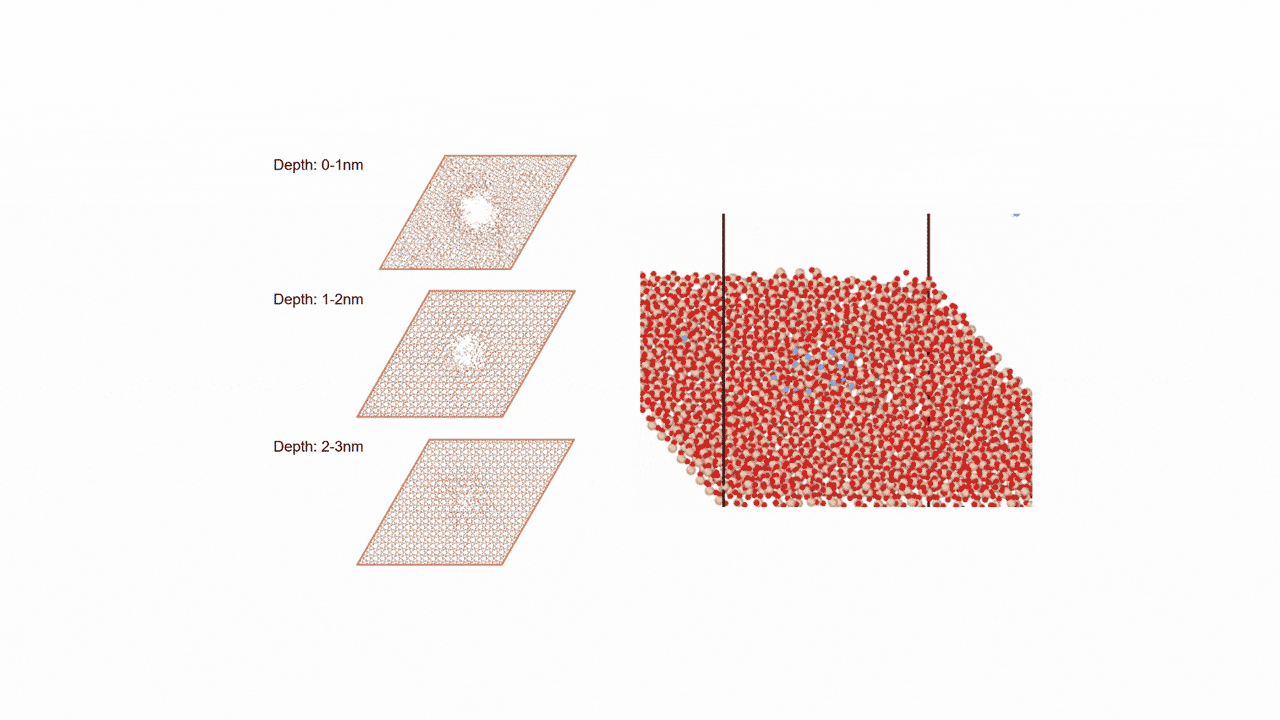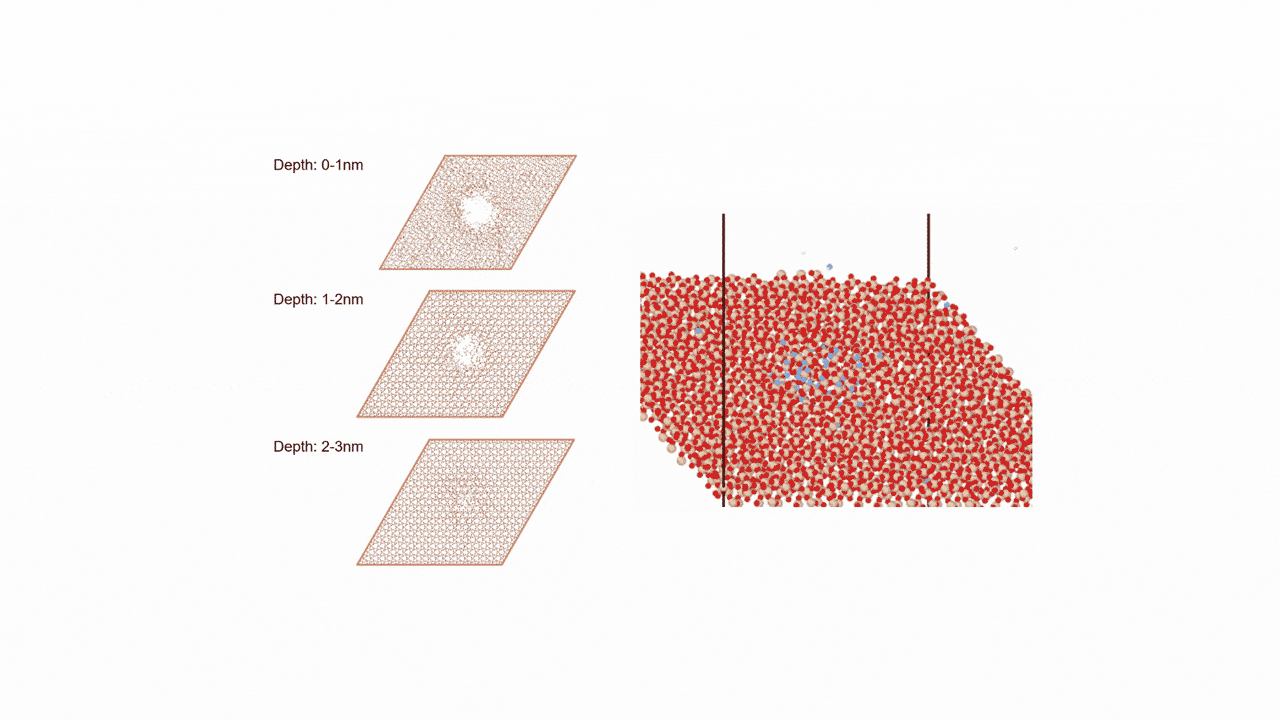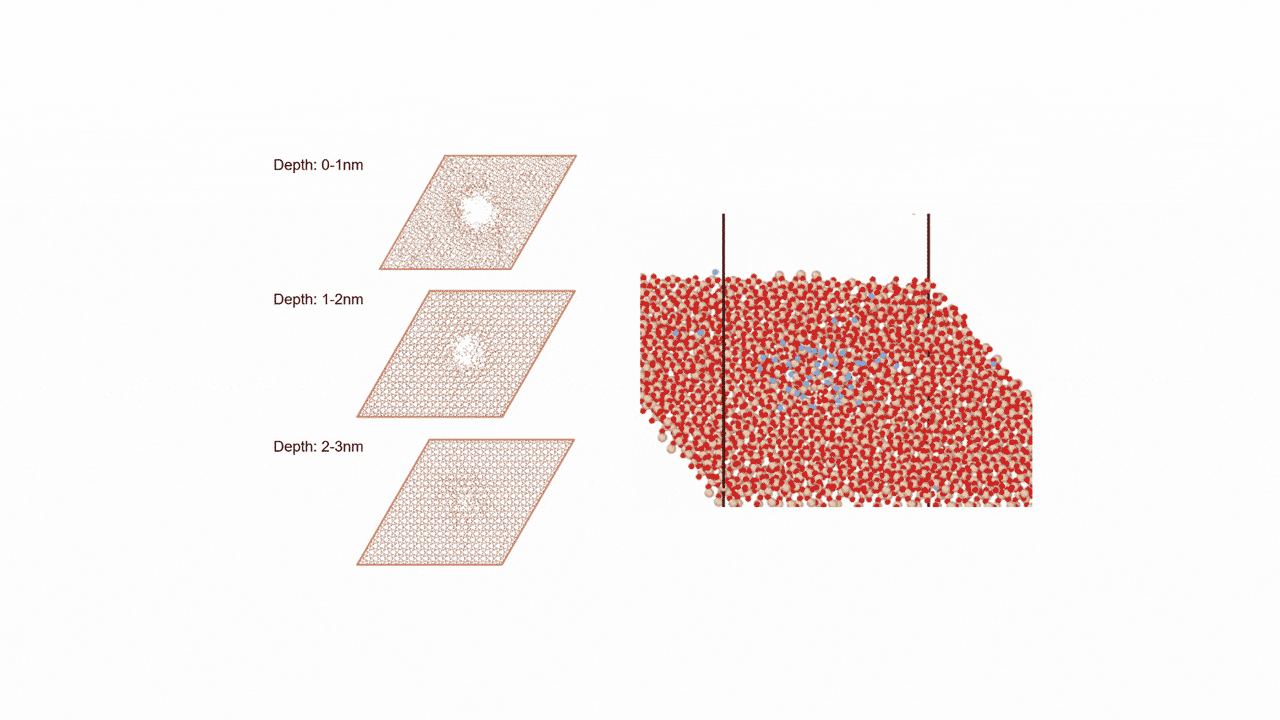
RWGS-CL反応雰囲気下で存在すると考えられるCu2Inの組成を持つ金属間化合物に対して、約2000原子からなる球状クラスター(半径19 Å, Cu1311In672)を作成しました。
実際の触媒上の担持金属はこのようなナノオーダーのクラスターで存在している一方、従来の密度汎関数理論(DFT)計算ではこれほど多くの原子数を持つモデルは計算コストが高く、MD計算を行うことは非常に困難でした。
本検討ではMatlantisでNosé-Hoover熱浴を用いたMD計算を行うことで、実際の反応温度である773 Kにおける金属間化合物クラスターモデルの表面状態を評価しました。
計算条件

計算結果と展望
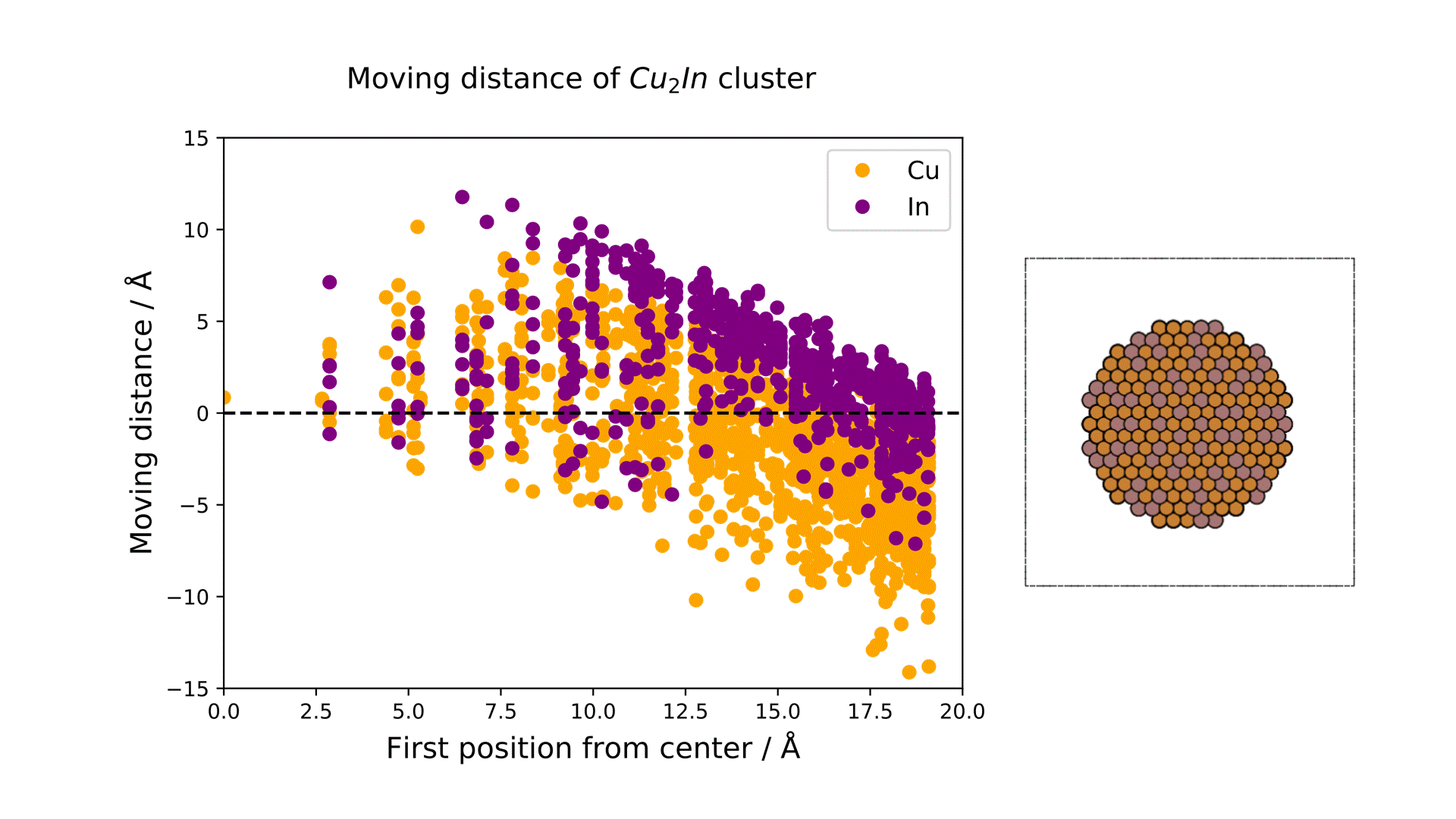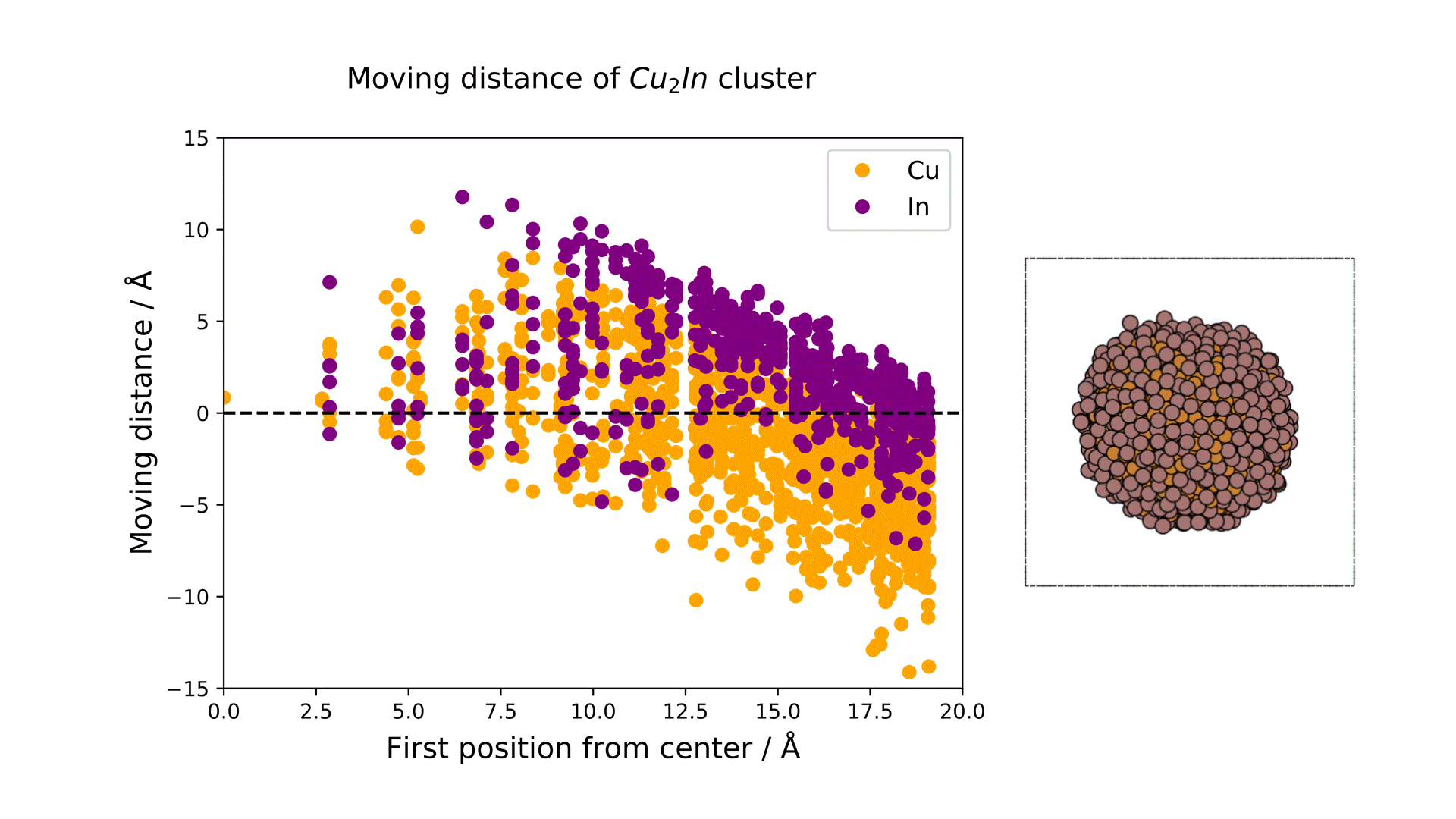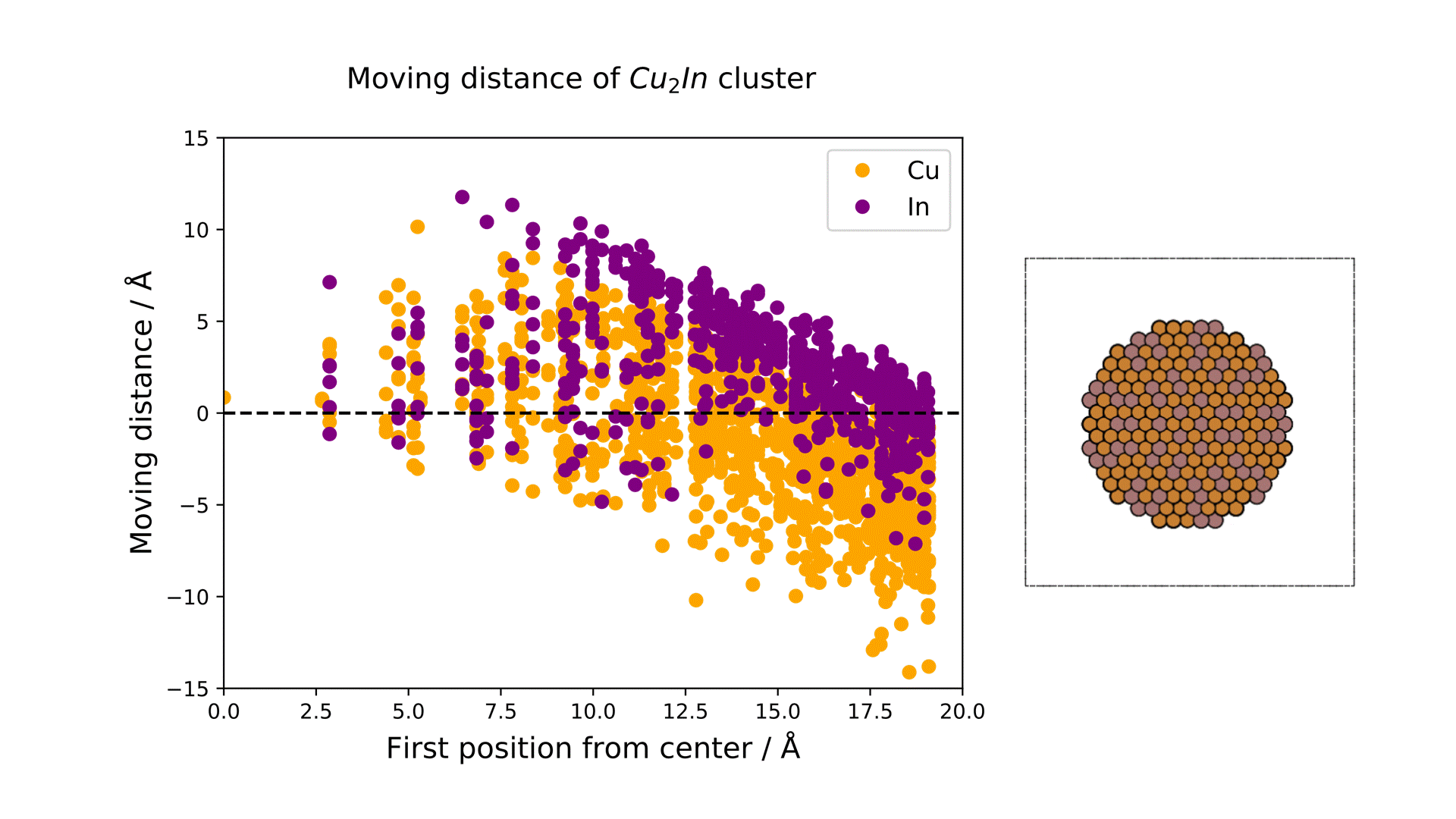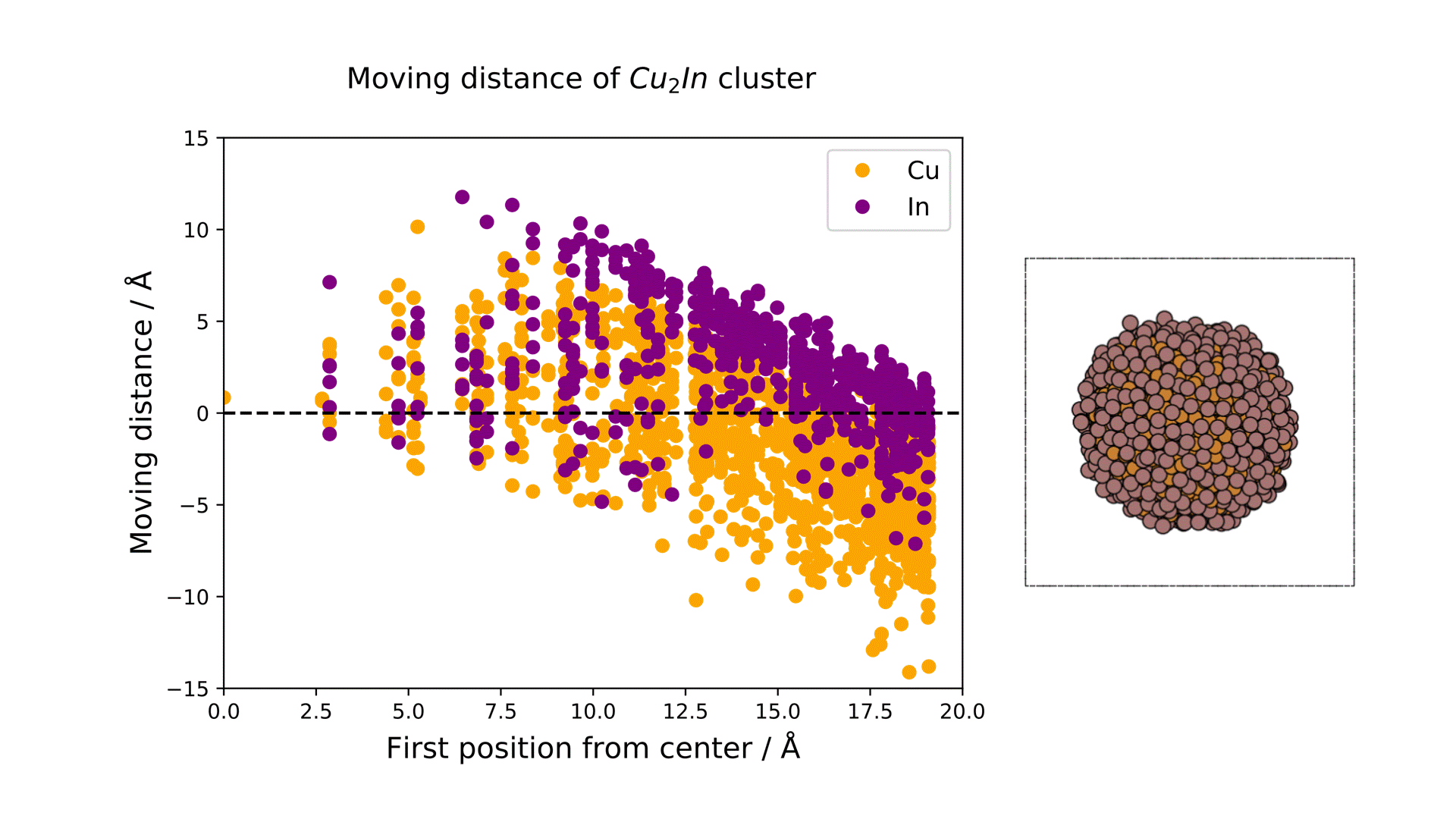
MD計算前後での各元素の変位を右図に示します。横軸は計算前の各原子のクラスターの中心からの距離を、縦軸は反応前後での各原子のクラスターの半径方向の移動距離を示しています。
結果から、クラスターの中心近くではCuもInも同様の変位挙動を示している一方、表面近くではInが外側に移動し、Cuは内側に移動するといった傾向がみられました。
これより、実温度でのCu-In金属間化合物は表面がInリッチな合金構造となっていることが予想され、Matlantisを用いることで現実に近いクラスターサイズを持つモデルの実反応温度での表面構造を低計算コストで評価することができました。
Matlantisは従来のDFT計算よりも多くの原子数を持つモデルを計算できるため、金属間化合物をIn2O3上に配置するなどして現実に近いモデルを構築することにより、実験条件における原子位置や反応性をシミュレートするという応用が期待されます。
また、Matlantisの汎化性を活かして、組成を変えた検討を網羅的に行うことにより、既報の材料より優れた材料を理論計算から予言できる可能性もあります。
参考文献
[1] IPCC AR6 Climate Change 2021: The Physical Science Basis. https://www.ipcc.ch/report/sixth-assessment-report-working-group-i/ [2] J.-I. Makiura, S. Kakihara, T. Higo, N. Ito, Y. Hirano, and Y. Sekine, Chem. Commun., 2022, 58, 4837-4840. https://pubs.rsc.org/en/content/articlelanding/2022/cc/d2cc00208f [3] J.-I. Makiura, T. Higo, Y. Kurosawa, K. Murakami, S. Ogo, H. Tsuneki, Y. Hashimoto, Y. Sato, and Y. Sekine, Chem. Sci., 2021, 12, 2108-2113. https://pubs.rsc.org/en/content/articlelanding/2021/sc/d0sc05340f事例提供者プロフィール
早稲田大学 関根研究室

タグ
本事例の公開日:2023.03.09
-




 URLを
URLを
コピーしました