2024.2.29
製品・サービス
PFPモデル v5.0.0リリース(メジャーアップデート)のお知らせ
概要
2021年7月のMatlantisのリリース以降、コア技術となるNNP (Neural Network Potential )であるPFP (Preferred Potential) [1] は約半年に1回のアップデートを行い、2024年2月現在では最新版となるv5.0.0が提供されています。
ここでは、最新版となるv5.0.0の検証結果について紹介いたします。
結果
Matbench Discoveryによるベンチマーク
Matbench Discovery [2] は、新しい安定な無機結晶を探索することを想定したベンチマークです。 Materials Project [3]の構造を元素置換することで生成した構造(WBM Dataset [4])を入力として、構造最適化を行い形成エネルギーなどを計算した結果を、DFT (Density Functional Theory) による結果と比較します。
注意点として、Matbench DiscoveryはAcからPuまでのアクチノイドの一部を対象としている一方で、PFPはこれらの元素に対応していません。そのため、これらの元素が含まれる構造は以下の結果から除外されています。また、Matbench DiscoveryのDFT条件とPFPデータセットのDFT条件には差異があります。この差異を吸収することを目的とし、補正を行っています。補正の詳細については[詳細]の章を参照してください。
Matbench Discoveryには、Materials Project [3] のデータセットを複数のアーキテクチャで学習した場合の結果がリーダーボードとして示されています。
Matbench Discoveryでは、ゼロ温度・ゼロ圧力での相図に着目します。相図において、熱力学的に安定な結晶構造群からなる凸包(convex hull)からのエネルギー差をenergy above hullと呼び、結晶構造の安定性の判定に用いられます。 安定性を判定する際のしきい値を変えながら、TPR (true positive ratio, 真陽性率)とFPR(false positive ratio, 偽陽性率)との関係を示す、ROC(Receiver Operating Characteristic)曲線を描いています。
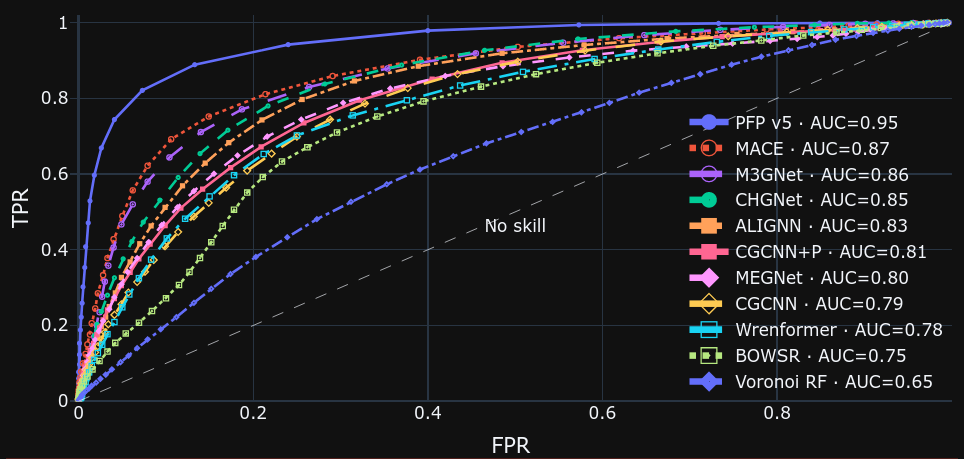
ROC曲線は、x=yの直線から離れて左上にいくほど、より優れた予測ができることを示しており、PFP v5.0.0が他のモデルと比較して優れていることがわかります。Materials Projectのデータセットと比べて、より多様で大規模なデータセットを用いて学習させていることがこの要因であると考えられます。
ある元素が含まれている構造に対するenergy above hullの元素毎のMAEを、PFP v5.0.0とリーダーボード上で最も優れているモデルであるMACEとで比較しました。
| PFP v5.0.0 | MACE |
 | 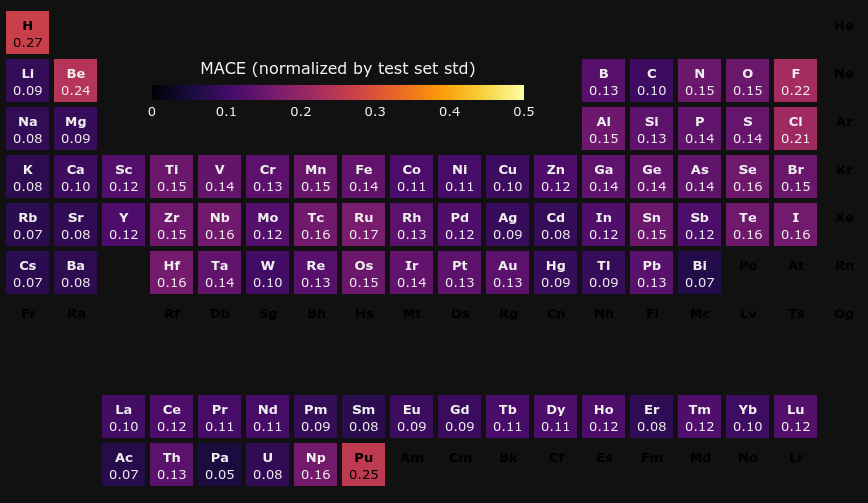 |
PFP v5.0.0では極端にMAEが大きい元素がなく、様々な元素に対応した汎用的なポテンシャルであると言えます。Mn, Fe, Crの遷移金属のMAEが比較的大きいですが、これは、これらのDFT計算がスピンなどにより難しいからであると考えています。 また、YbはMACEよりMAEが大きいですが、これはYbのpseudo potentialが、PFPデータセットとMaterials Projectとで異なるものを選択しているからであると考えられます。
Matbench Discoveryで示されている他の指標を以下に示します。 指標の定義は[2]を参照してください。 いずれも、PFP v5.0.0は高いスコアを示しており、新しい安定な無機結晶を探索するというタスクにおいて優れていると言えます。他モデルの値は2024年2月14日の公式Webページのリーダーボードの値を参照しています。
| F1 ↑ | DAF ↑ | Prec ↑ | Acc ↑ | TPR ↑ | TNR ↑ | MAE ↓ | RMSE ↓ | R2 ↑ | |
| PFP v5.0.0 | 0.76 | 5.26 | 0.75 | 0.92 | 0.77 | 0.95 | 0.03 | 0.07 | 0.84 |
| PFP v5.0.0 (72elem) | 0.77 | 8.16 | 0.76 | 0.93 | 0.79 | 0.96 | 0.03 | 0.07 | 0.85 |
| MACE | 0.674 | 3.1378 | 0.584 | 0.885 | 0.80 | 0.896 | 0.06 | 0.10 | 0.6770 |
| CHGNet | 0.61 | 3.3609 | 0.512 | 0.854 | 0.764 | 0.876 | 0.06 | 0.10 | 0.69 |
| M3GNet | 0.57 | 2.8867 | 0.445 | 0.810 | 0.8077 | 0.81 | 0.07 | 0.121 | 0.5860 |
PFP v5.0.0 (72elem) は、PFPが対応している72元素に絞ったときのスコアです。
表内の指標について個別に説明します。表の右側のMAE, RMSE, R^2といった指標は回帰誤差に紐づく量で、全体的なずれが少ないほど良い評価になります。PFPは特にこれらの指標について良いスコアを示しており、PFPの汎用ポテンシャルとしての性質が現れたものと考えられます。
F1およびPrec, Acc, TPR, TNRは結晶構造のエネルギーの値が凸包に対して正か負かの二値分類に基づくスコアです。PFPはこれらのスコアについても良い値を示しています。一方でデータ点の多くが凸包のごく近傍にあるため、比較的学習元のデータセットのDFT計算条件等によって影響を受ける繊細なものと考えられます。
DAFは計算できていない構造が含まれると数値が高くなる傾向にあるため、ここでは説明を割愛します。
結晶体積の再現性検証
PFP v4.0.0では、有機結晶の体積をDFT計算と比較して過剰に評価していたという課題がありました。しかし、PFP v5.0.0ではこの課題が大幅に改善されました。
図にはCOD (Crystallography Open Database) [5] から取得した構造を対象として、DFTとPFPで計算した体積の比較結果を示しています。有機結晶や錯体結晶の体積には分散力が大きな影響を及ぼすことが知られており、GerminらのD3補正[6,7]を加えています。無機結晶に関してはD3補正を加えていません。
| 相対体積 | 相対誤差 | |
| 有機結晶 | 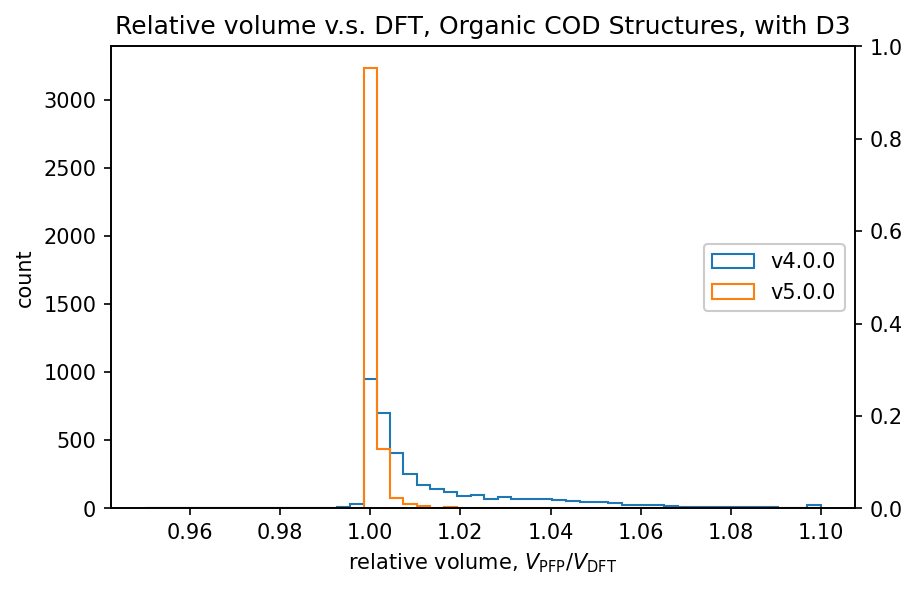 | 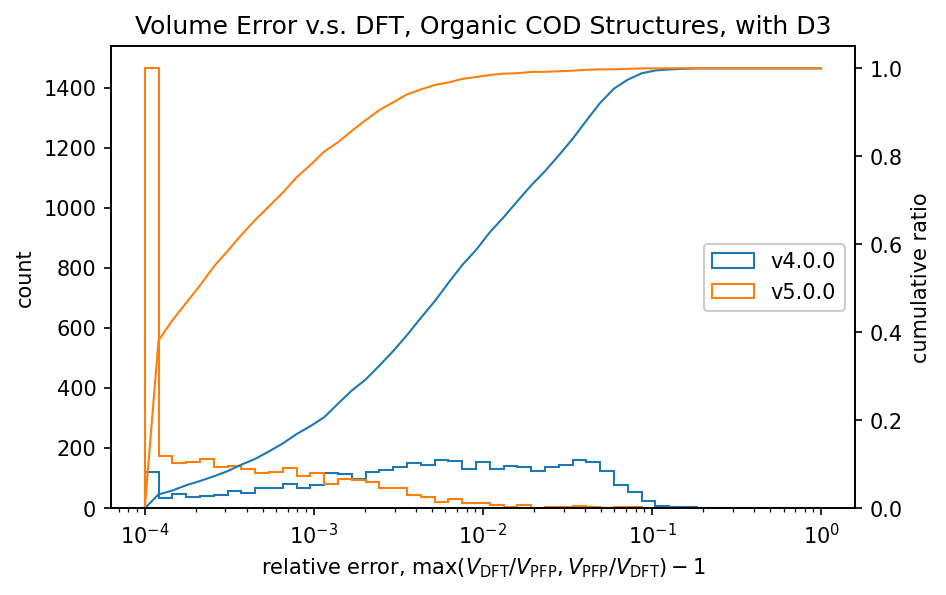 |
| 錯体結晶 | 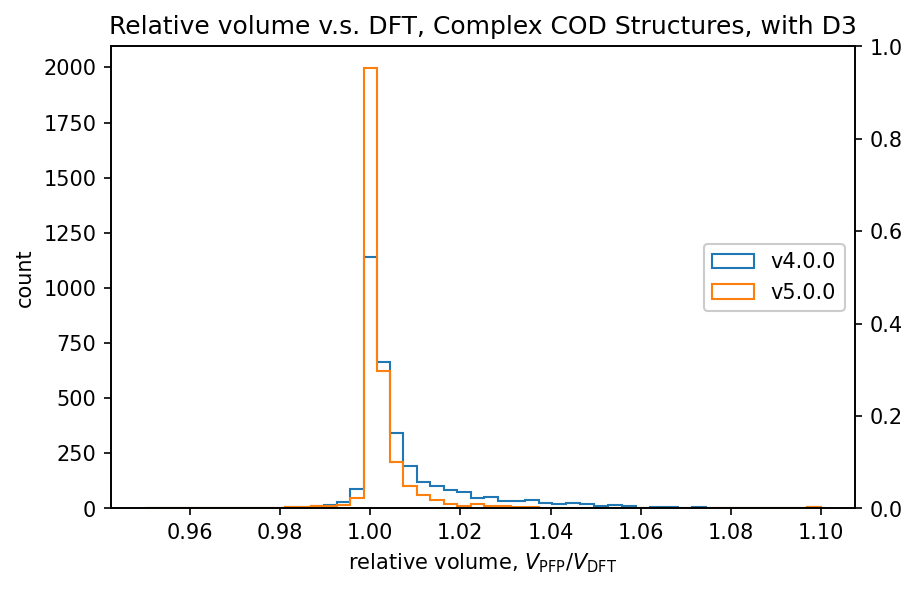 | 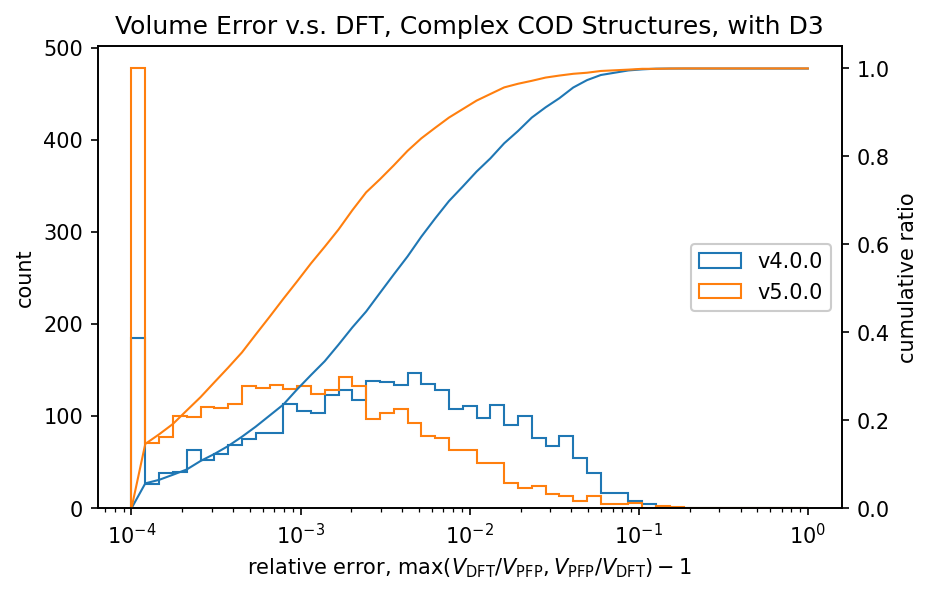 |
| 無機結晶 | 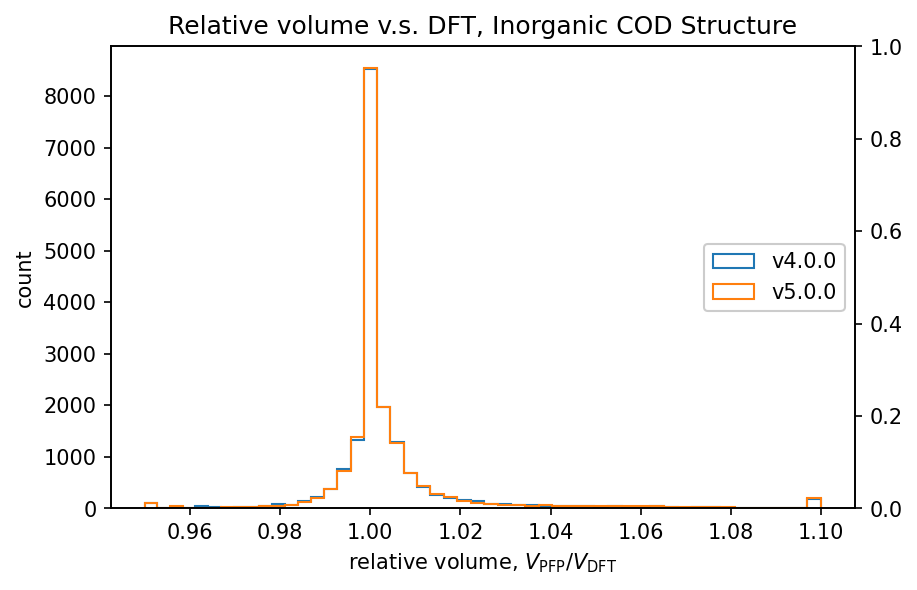 | 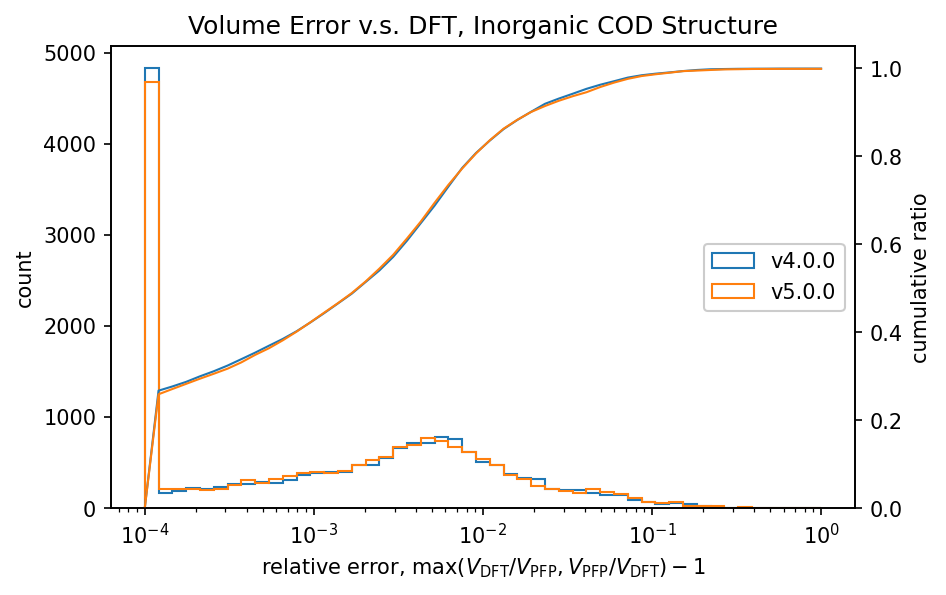 |
有機結晶に対する相対体積のヒストグラムから、PFP v4.0.0はDFTに対して体積を過剰に評価している傾向がありましたが、PFP v5.0.0ではこの傾向が大幅に改善されていることがわかります。また、PFP v5.0.0では、90%の有機結晶構造の体積を、相対誤差0.23%以内の高い精度で求めることができました。一方、PFP v4.0.0では相対誤差が4.4%以内であったため、v5.0.0で大幅に改善されています。錯体結晶に関しても、有機結晶ほどの改善は見られませんが、PFP v5.0.0ではv4.0.0と比較して体積の再現性が向上しています。無機結晶に関しては再現性は同等と言えます。
PFP v5.0.0では、データセットの拡充により、有機結晶や錯体結晶でのポテンシャルエネルギー曲面の再現性が大幅に改善されています。これが有機結晶や錯体結晶の体積の再現性の向上につながったと考えられます。前述のMatbench Discoveryは主に無機結晶を対象としたものですが、これらの結果から、PFPは有機結晶や錯体結晶に対する再現性も高い、汎用的なポテンシャルであると言えます。
まとめ
- Matbench Discoveryによるベンチマークの結果から、PFP v5.0.0が既存のモデルと比較して、新しい安定結晶を探索するというタスクにおいて優れていることが示されました。より多様で大規模なデータセットによって学習させていることがその要因と考えられます。
- PFP v5.0.0では、PFP v4.0.0と比較して、有機結晶と錯体結晶の再現性が向上しました。データセットの拡充などの影響によると考えており、特に有機結晶の再現性の向上が顕著です。また、PFPはMatbench Discoveryが対象とする無機結晶だけでなく、有機結晶や錯体結晶に対する再現性も高い汎用的なポテンシャルであると言えます。
詳細
ここでは、計算手法の詳細や追加の情報を示します。
Matbench Discoveryによるベンチマーク
Matbench DiscoveryのDFT条件はMaterials Projectのものを採用しています。PFPデータセットと概ね同じですが、smearing手法などが異なります。 ここではDFT条件間の差異を吸収することを目的とし、Matbench DiscoveryのWBMデータセットと同様のDFT条件で作成されたMaterials Projectのデータセット( 2023-02-07-mp-computed-structure-entries.json.gz )を使い、このデータセットに対して形成エネルギーが合うように各元素の単体のエネルギーを最小二乗フィッティングしました。このデータセットはMatbench Discoveryベンチマークにおける学習用データセットであり、この処理によってテストデータの情報がリークすることはありません。
結晶体積の再現性検証
比較対象とするDFT計算は、VASP 5.4.4あるいはVASP 6.4.0を用いて行いました。VASP 5.4.4とVASP 6.4.0による結果の差異は十分に小さく、無視できることを確認しています。PFPの教師データ生成に用いている計算条件と同じ計算条件を用いており、PBE汎関数とPAW(Projector Augmented Wave)法を利用して計算しています。詳細についてはPFPの論文[1]を参照してください。なお、PFP v2からHubbard補正を行わない計算モードを提供しており、本検証はこれを対象としています。このため、比較対象とするDFT計算においても、Hubbard補正を行っていません。
結晶構造はCODから、PFP v3以降でサポートする72元素のみで構成される結晶構造のうち、単位胞の体積が2200 ų以下のものを取得しました。ただし、各サイトの占有率が0.99未満のものや、対称性の指定に問題があるもの等は除外しています。 有機結晶、錯体結晶、無機結晶は以下の定義に基づき分類しました。
有機結晶: H, C, N, O, P, S, F, Cl, Br, I のみで構成される構造
錯体結晶: 以下のすべての条件を満たす構造
- H, C, N, O, P, S, F, Cl, Br, Iのうちの2元素以上を含むこと
- H, C, N, O, P, S, F, Cl, Br, I以外の元素を1元素以上含むこと
- H, C, N, O, P, S, F, Cl, Br, Iで原子数の8割以上を占めること
. 無機結晶: 有機結晶と錯体結晶のいずれにも分類されない構造
これらの結晶構造に対して、原子にかかる力が0.03 eV/Å以下になるようにセルパラメータを含めた構造最適化をDFT計算で行い、ベンチマークデータ・セットを生成しました。先に述べたとおり、有機結晶と錯体結晶はD3補正を加えて構造最適化を行っています。
PFPでの構造最適化は、DFTによる構造最適化を行った構造を初期構造とし、原子にかかる力が0.03 eV/Å以下になるようにセルパラメータを含めた構造最適化をPFPで行いました。特記すべきこととして、有機結晶では、local minimumが数多く存在すると考えられます。別のlocal minimum同士の体積を比較することを極力避けるため、DFTによる構造最適化を行った構造を初期構造としています。
結晶構造のエネルギー・力の検証
有機結晶や錯体結晶の体積再現性が向上した理由として、これらの構造に対するポテンシャルエネルギー曲面の再現性向上が寄与していると考えております。以下に、エネルギーと力の再現性を評価した結果を示します。
上記のCODの結晶構造に対して、原子にかかる力が0.03 eV/Å以下になるようにセルパラメータを含めたDFT計算により構造最適化を行った後、原子位置に微小変位を加えた構造を生成しました。詳しくは、PFP論文[1]のSupporting Information, NOTE 10にある“site position displacement”を参照ください。ただし、変位の大きさは論文中よりも小さくしています。体積の再現性の検証とは異なり、ここでは構造最適化を含めて、D3補正を加えていないことに注意してください。
下図では、微小変位を加えた有機結晶構造に対するDFTとPFPでの一点計算の結果を比較しています。v5.0.0ではv4.0.0と比較しMAEが1/3程度となり、有機結晶のエネルギーと力の再現性が大きく向上していることがわかります。
| v5.0.0 | v4.0.0 | histogram | |
| エネルギー | 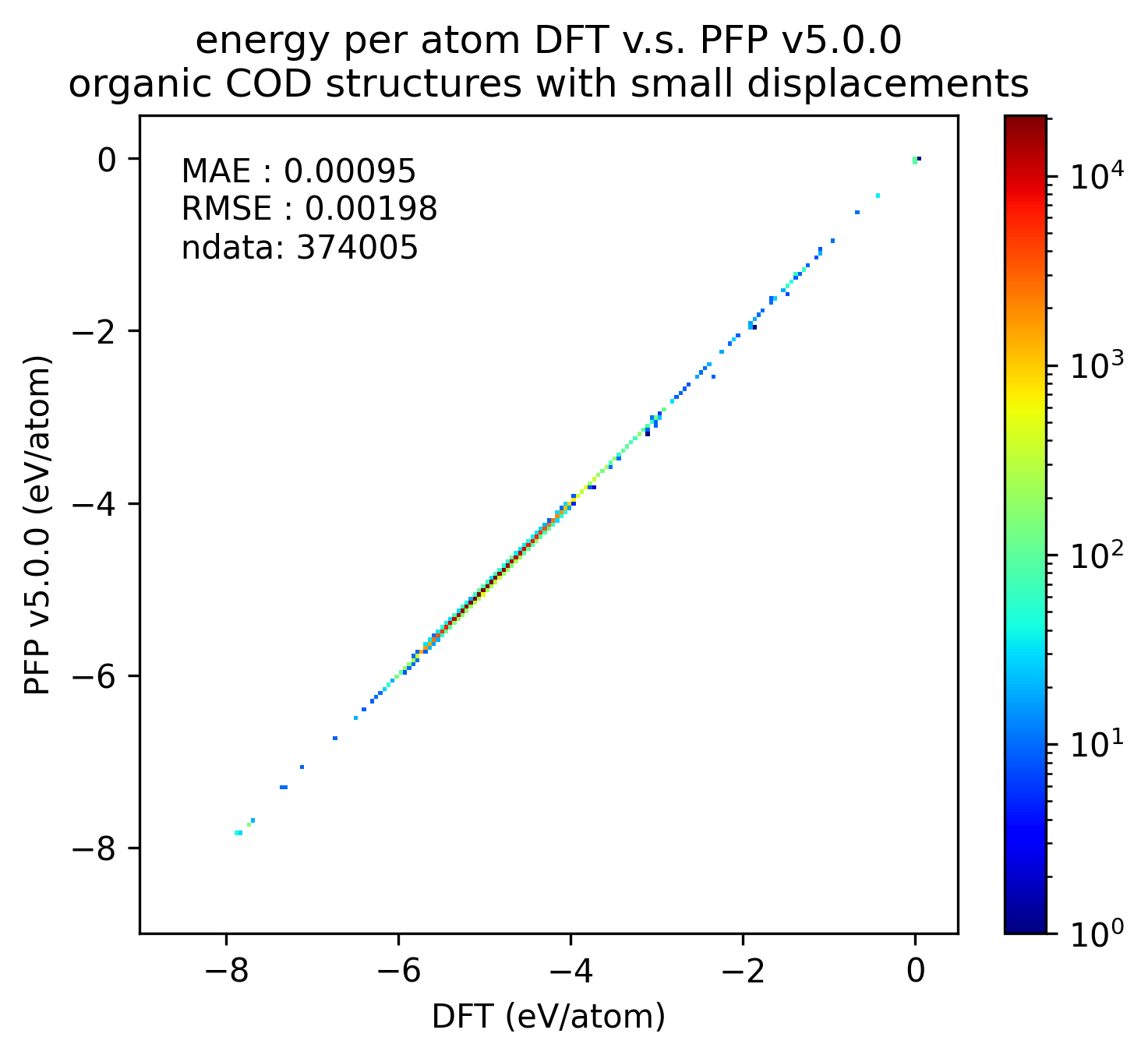 | 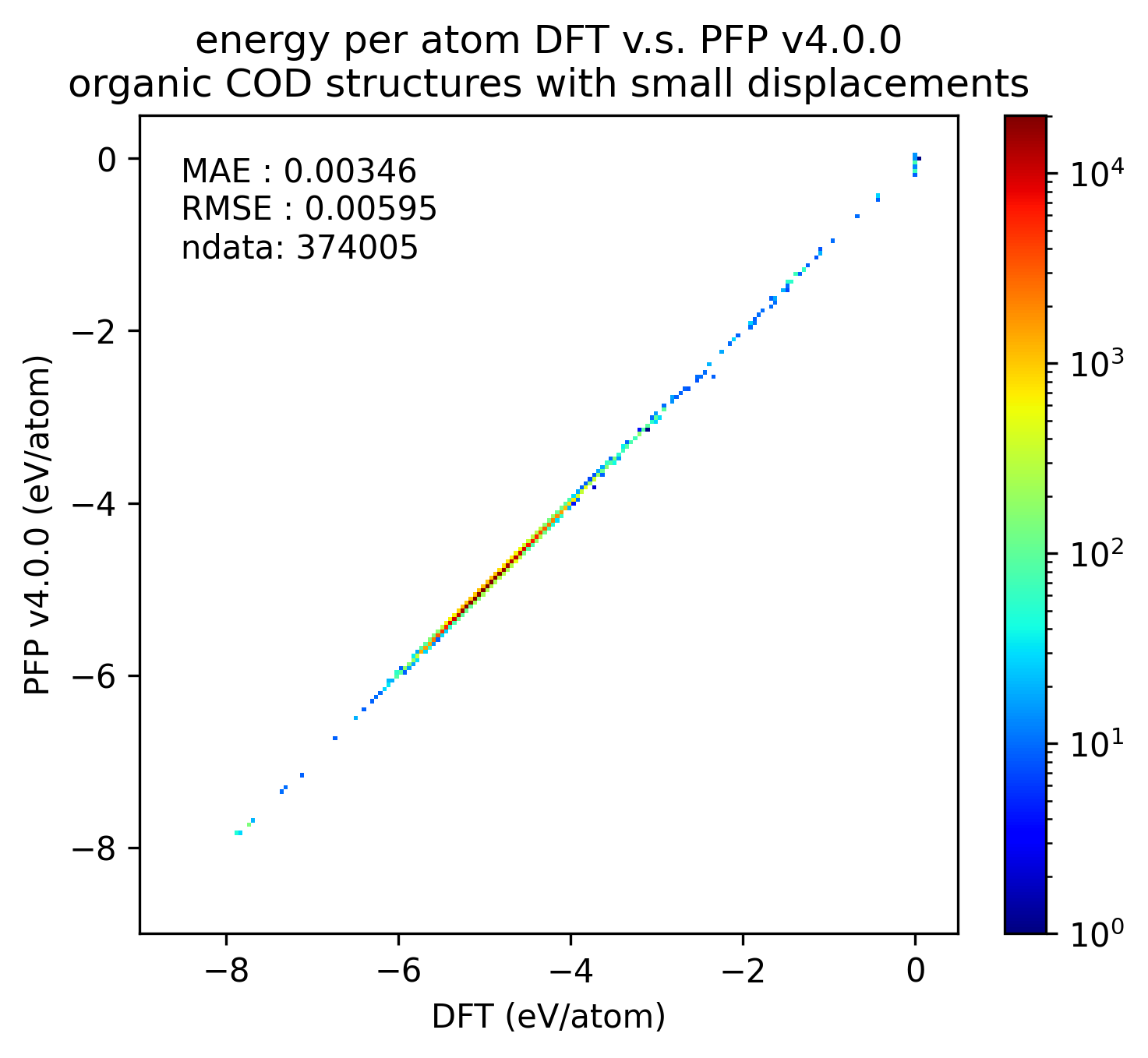 | 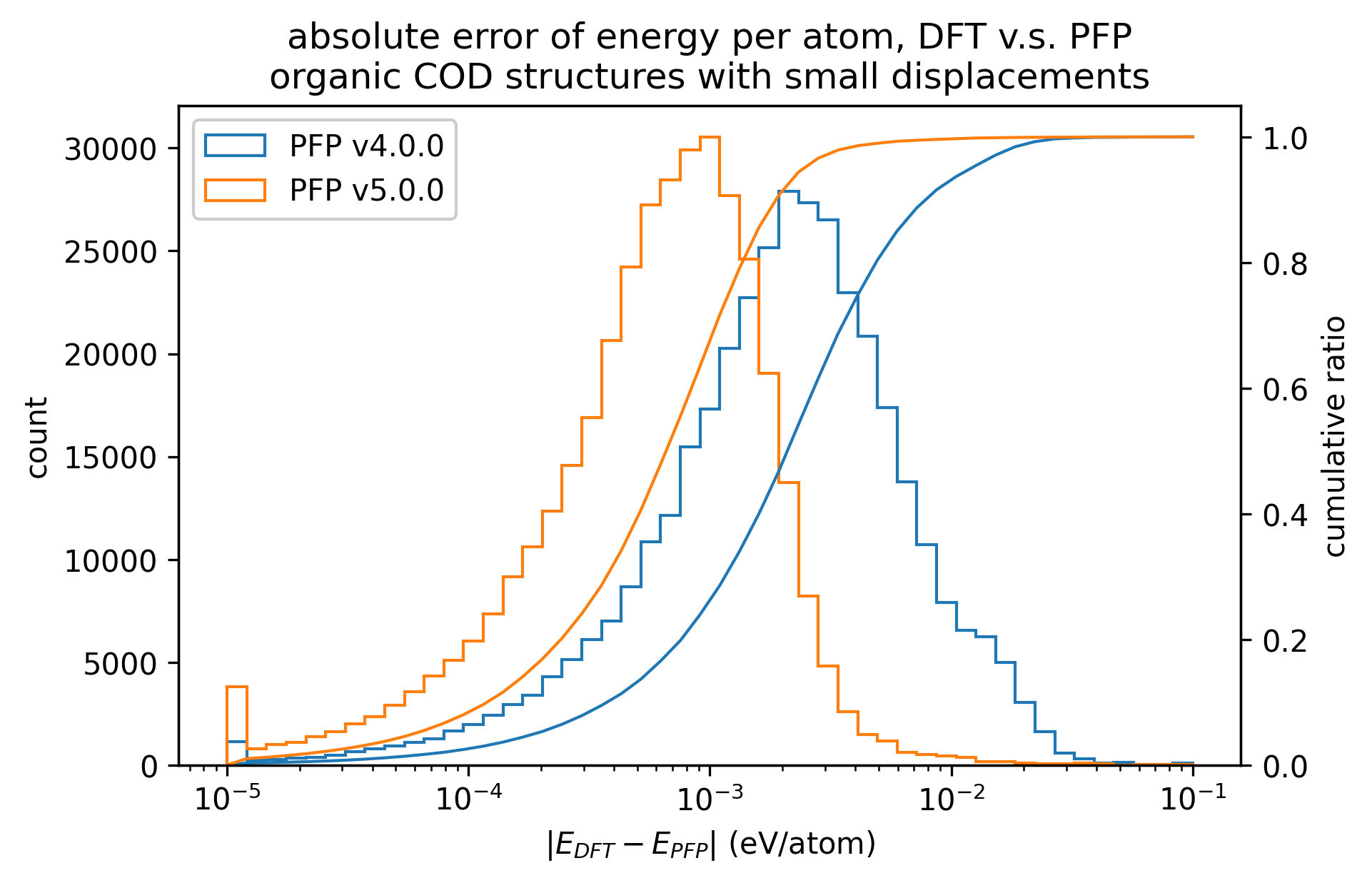 |
| 力 | 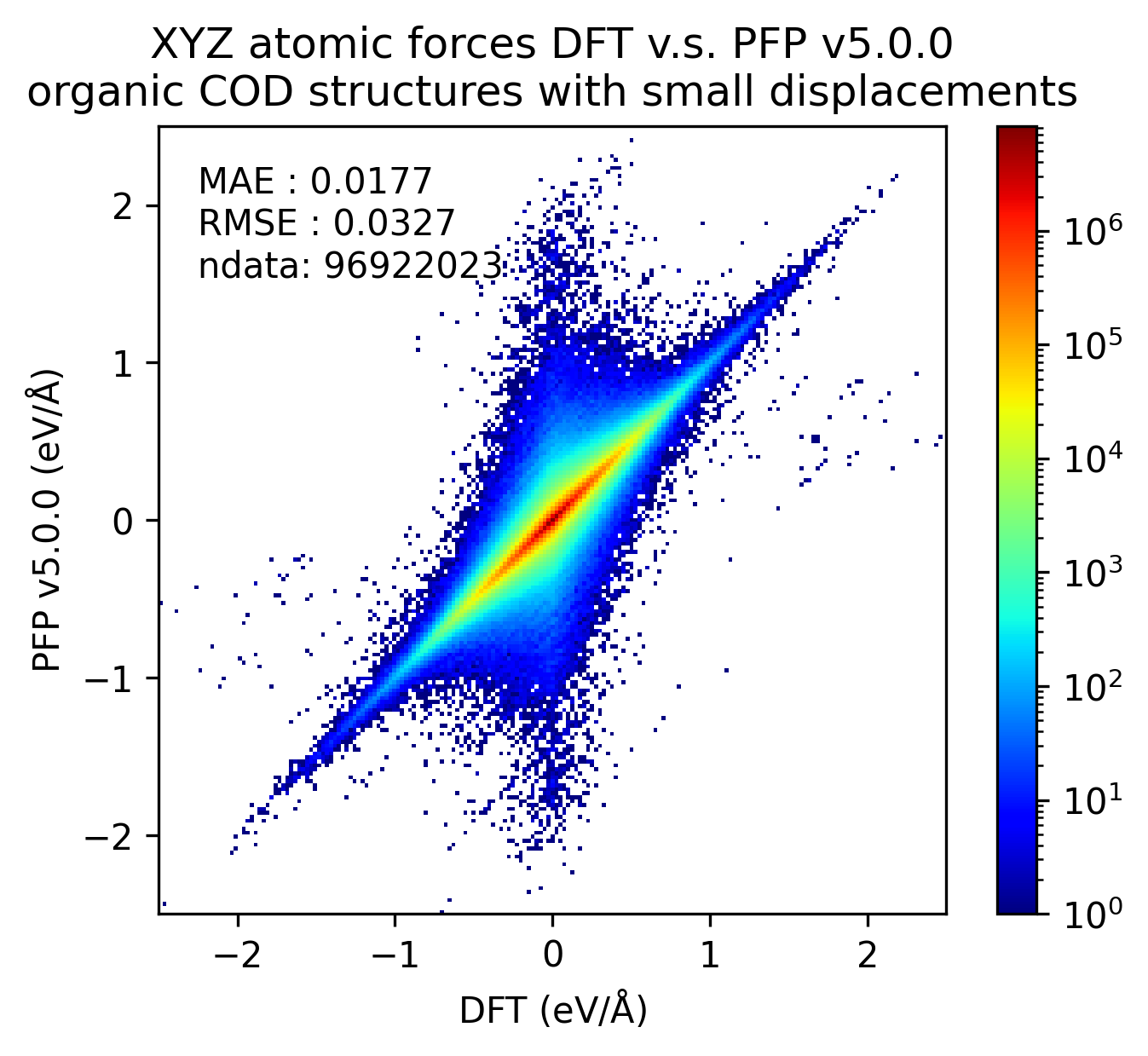 | 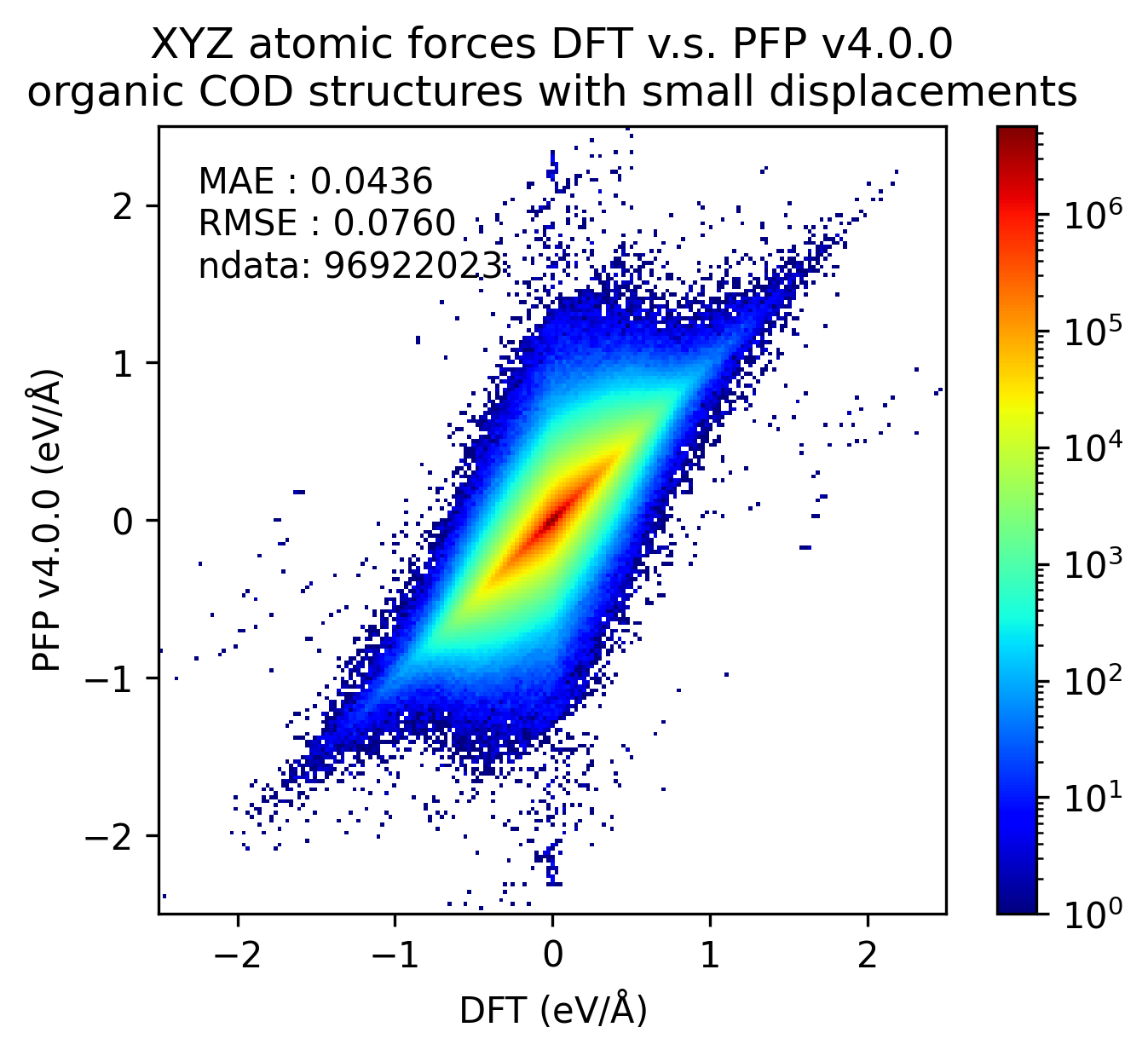 | 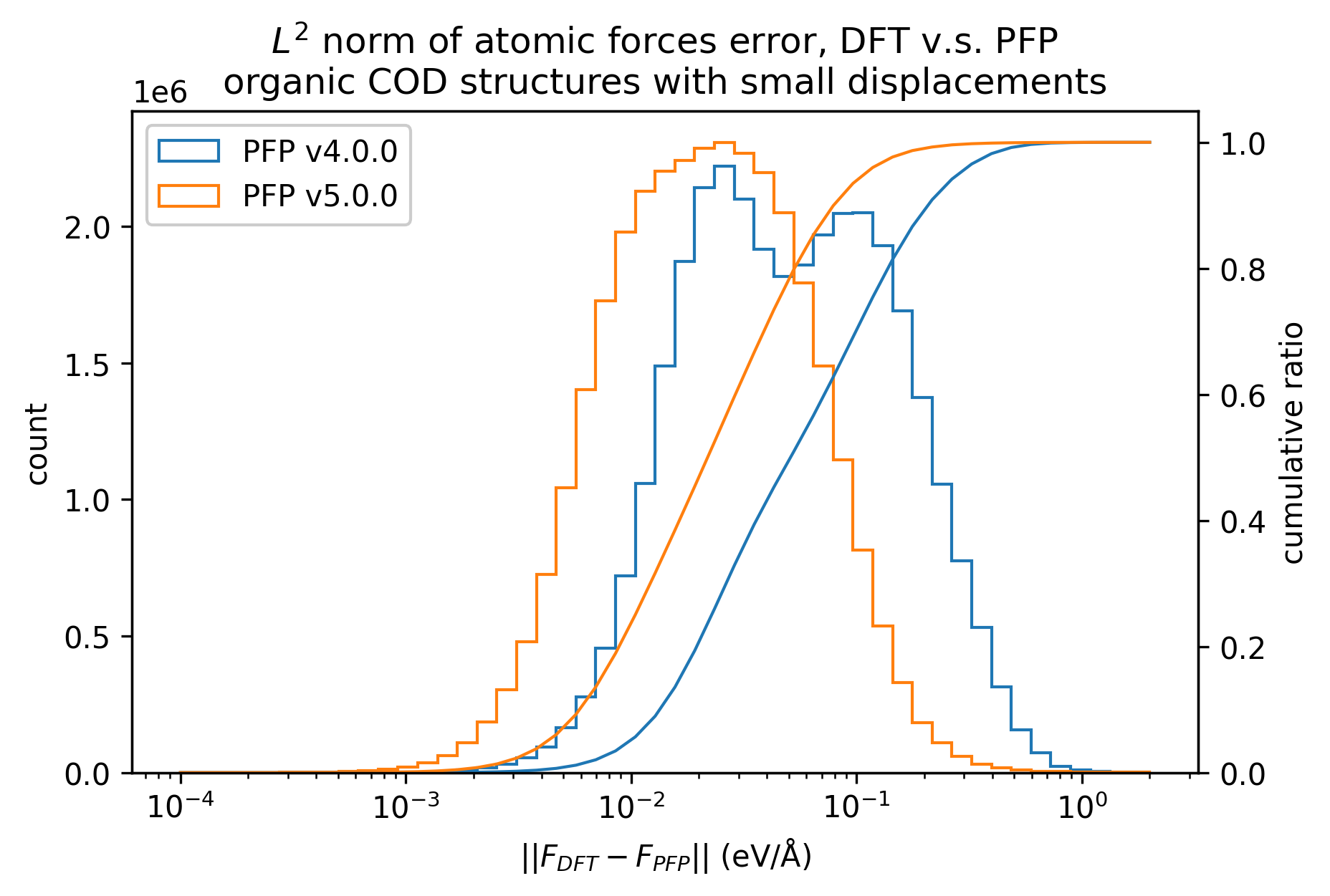 |
同様に、錯体結晶のエネルギーと力の再現性を示します。
有機結晶に対する結果ほど顕著ではないものの、v5.0.0ではv4.0.0と比較して錯体結晶のエネルギー・力の再現性が向上していることがわかります。
| v5.0.0 | v4.0.0 | histogram | |
| エネルギー | 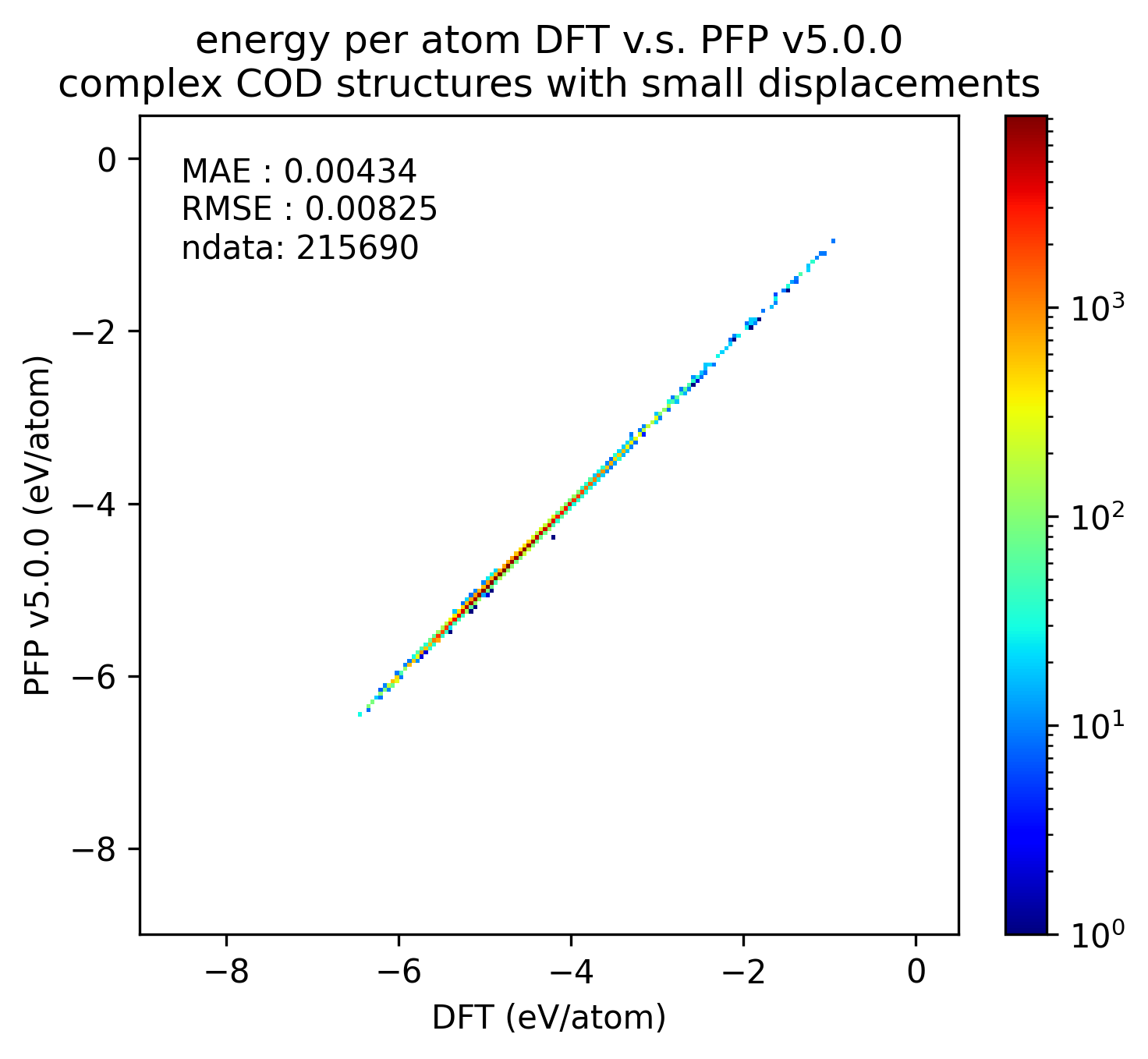 | 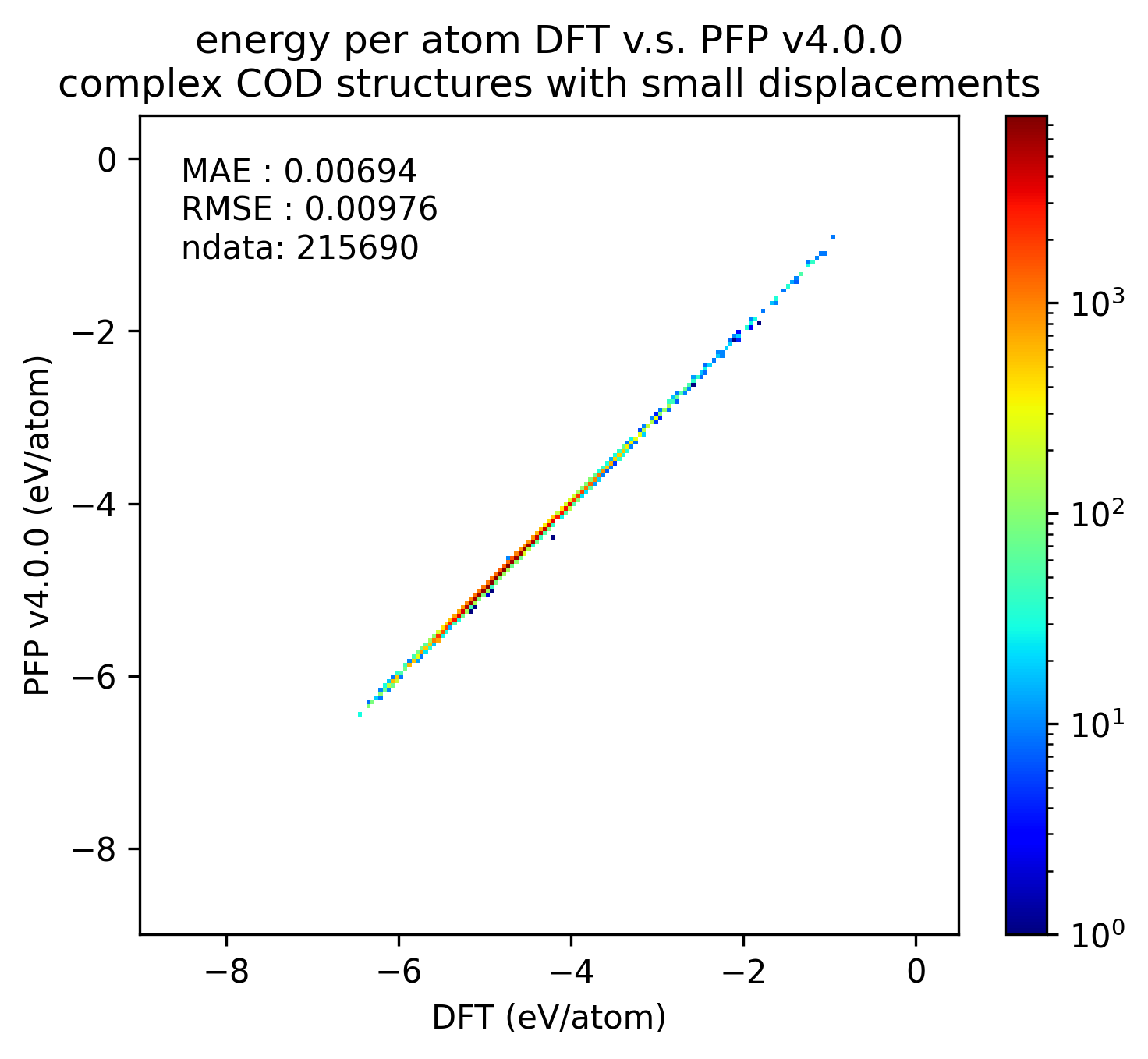 | 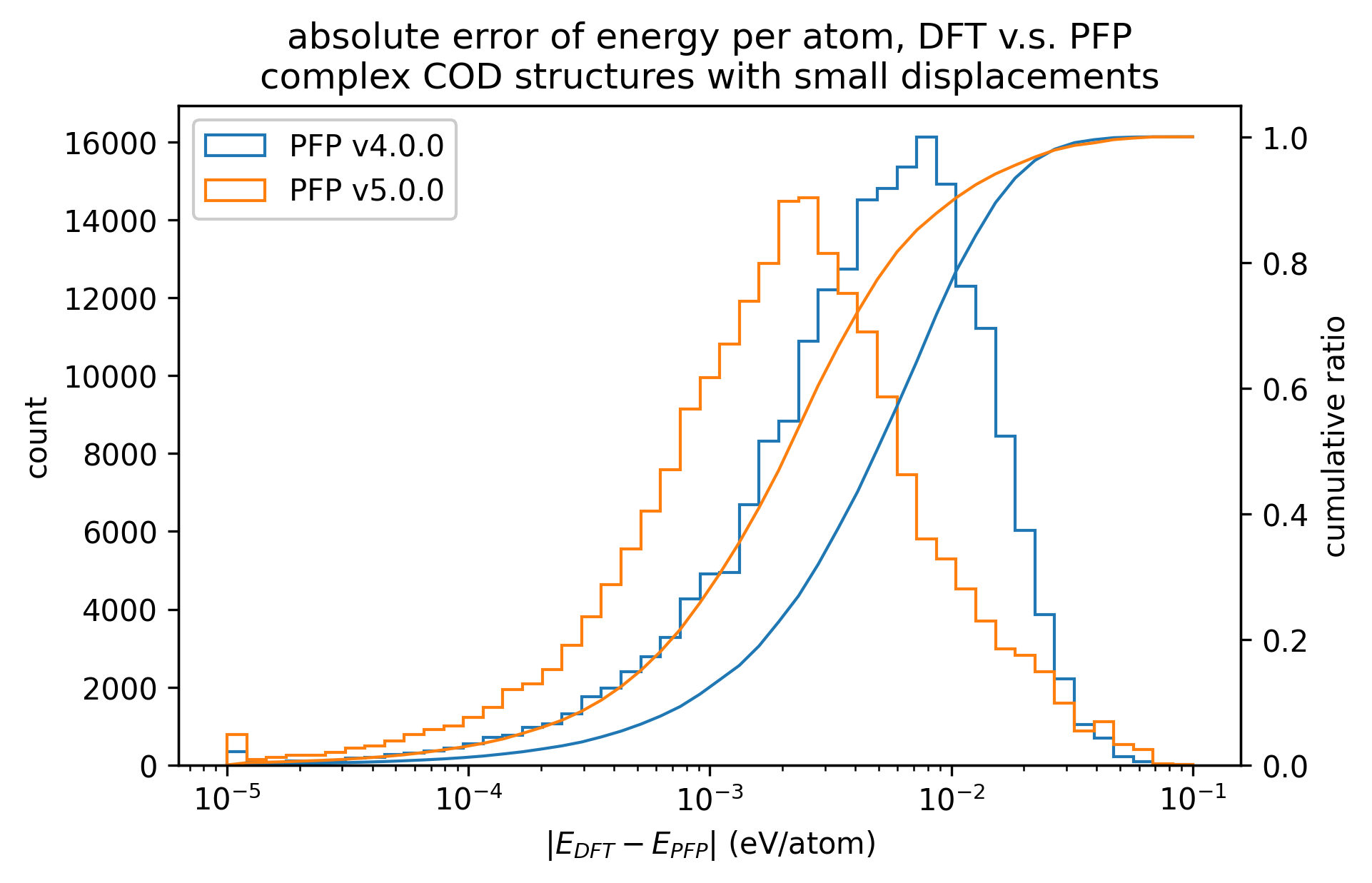 |
| 力 | 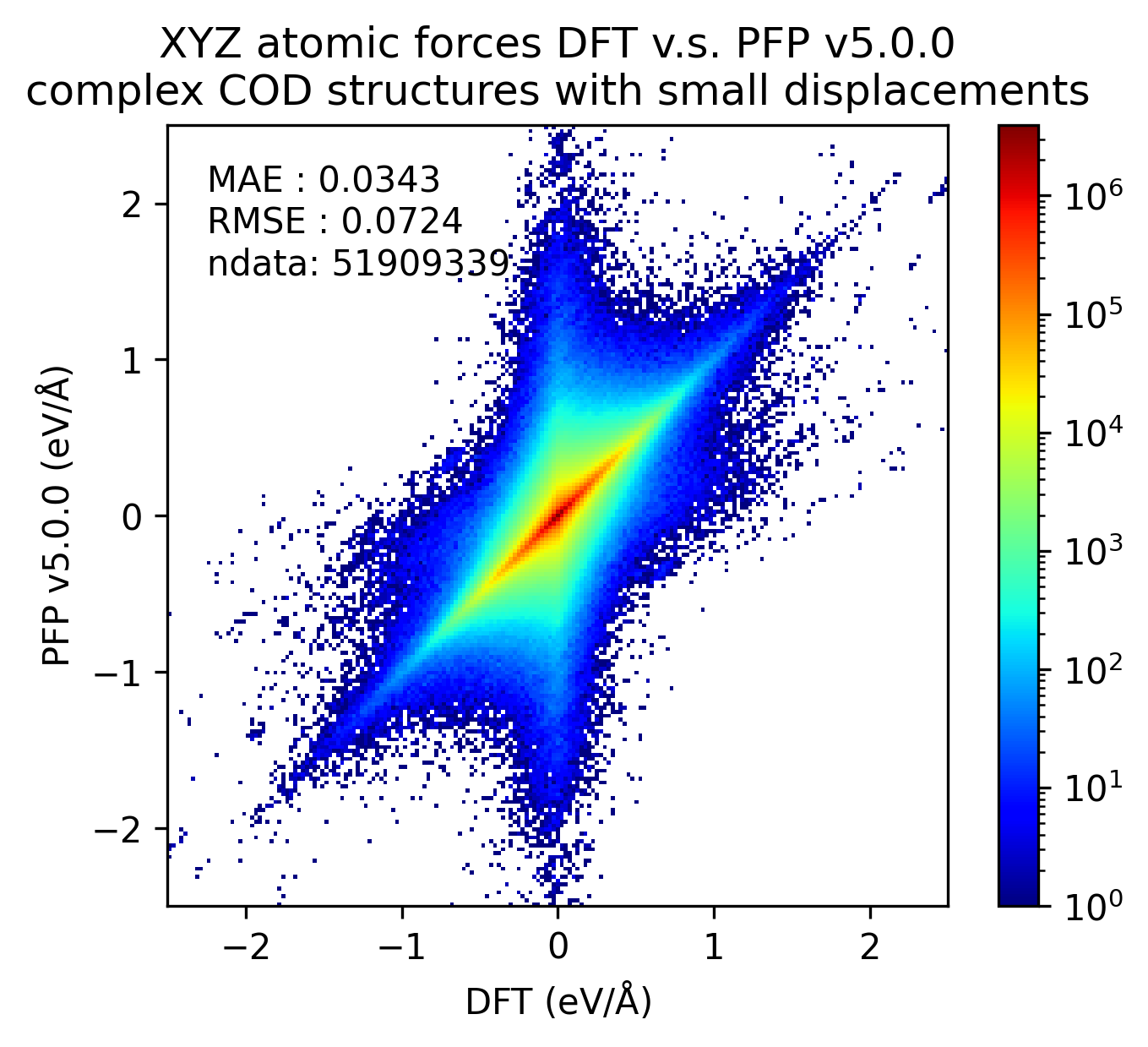 | 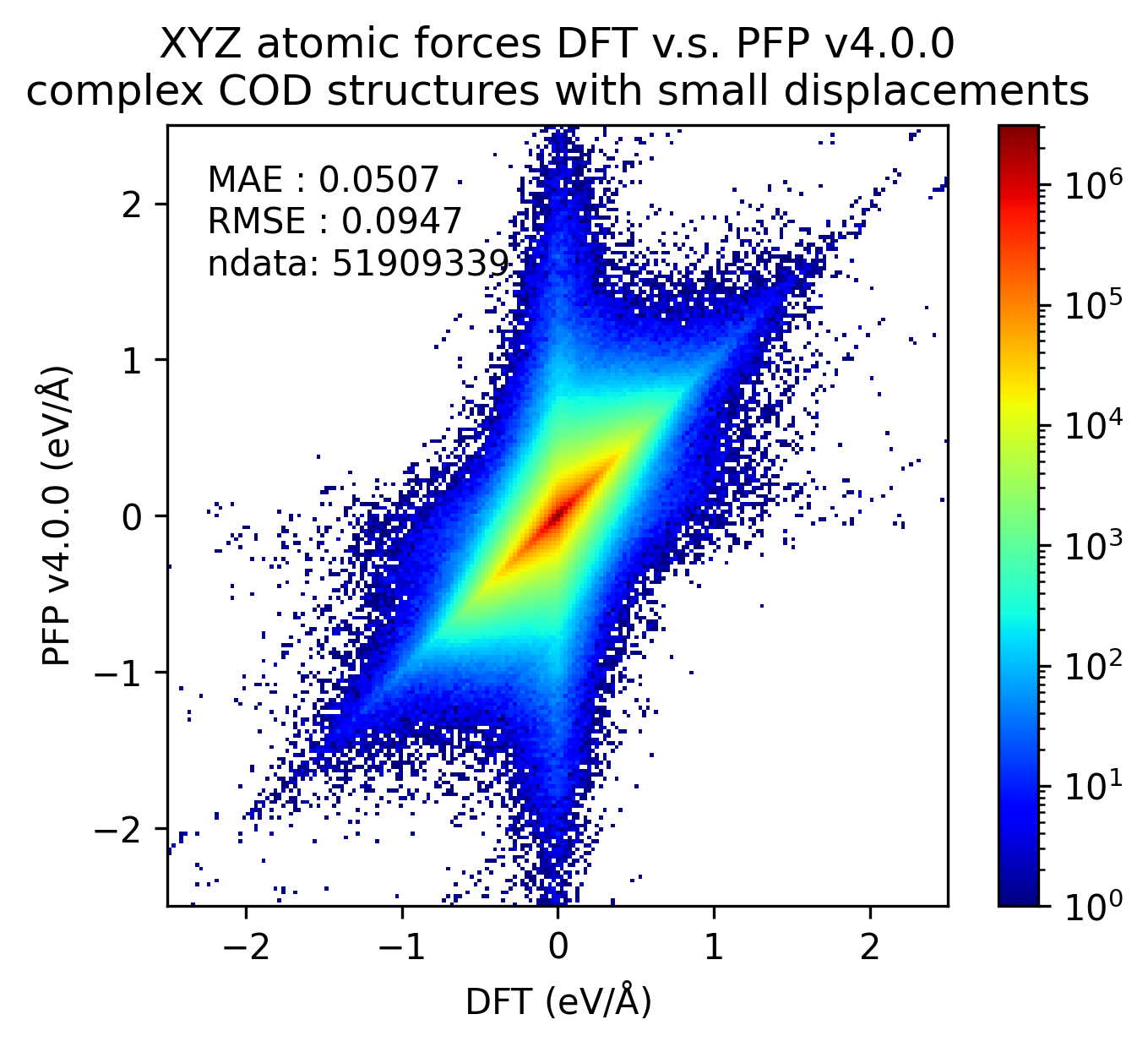 | 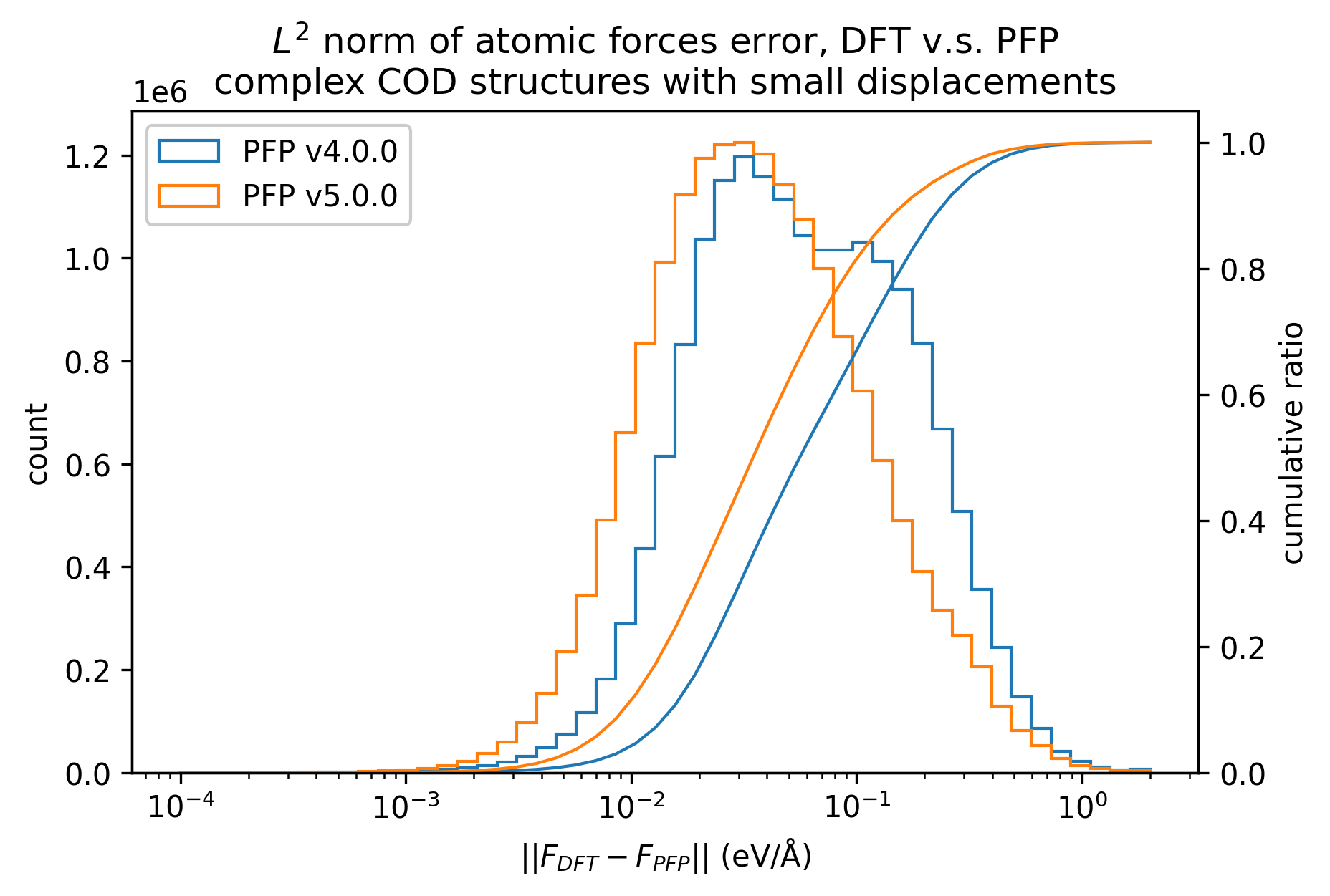 |
無機結晶のエネルギーと力の再現性を示します。MAEはPFP v5.0.0でわずかに改善しているものの、同等の再現性であると言えます。
| v5.0.0 | v4.0.0 | histogram | |
| エネルギー | 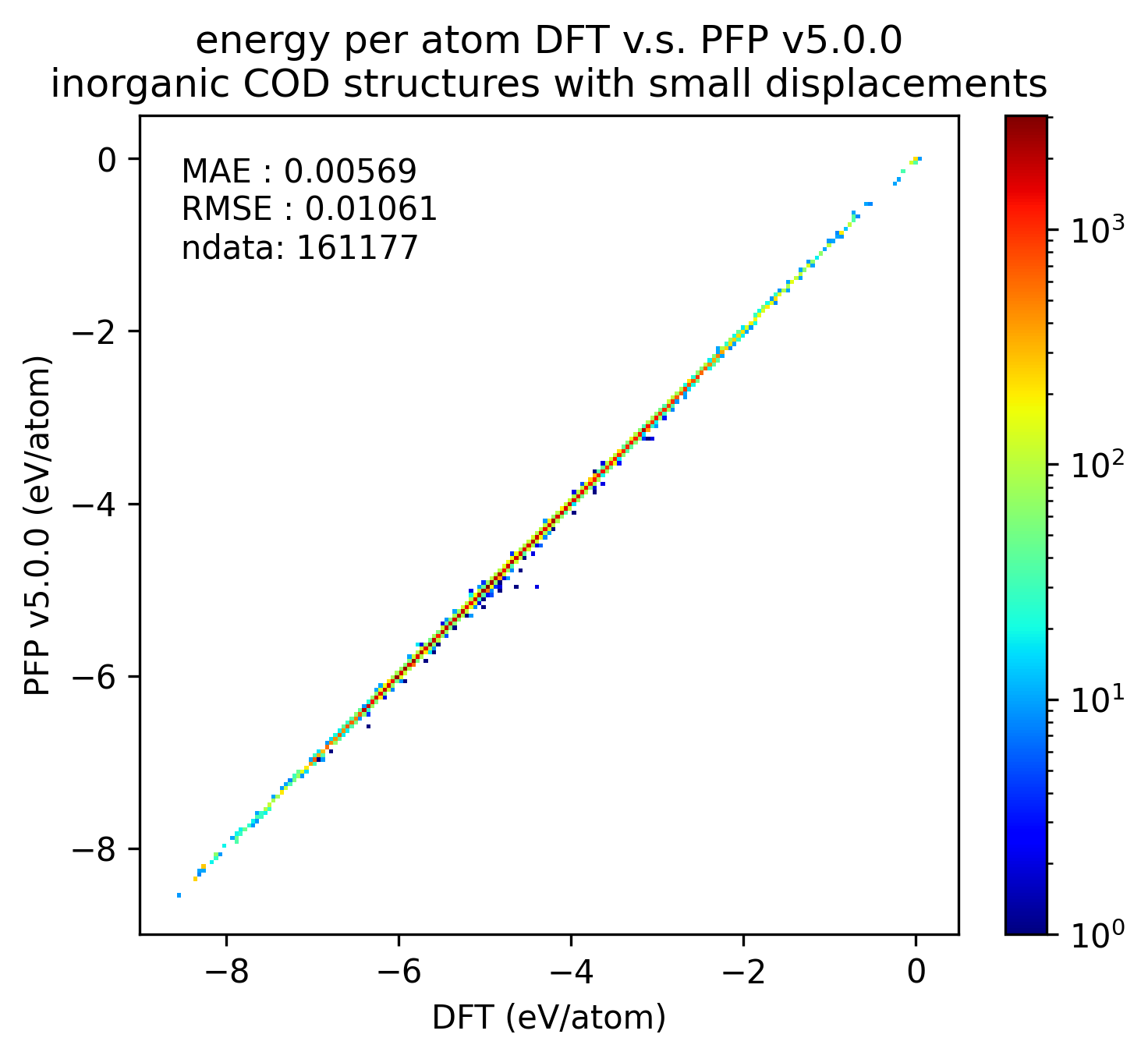 | 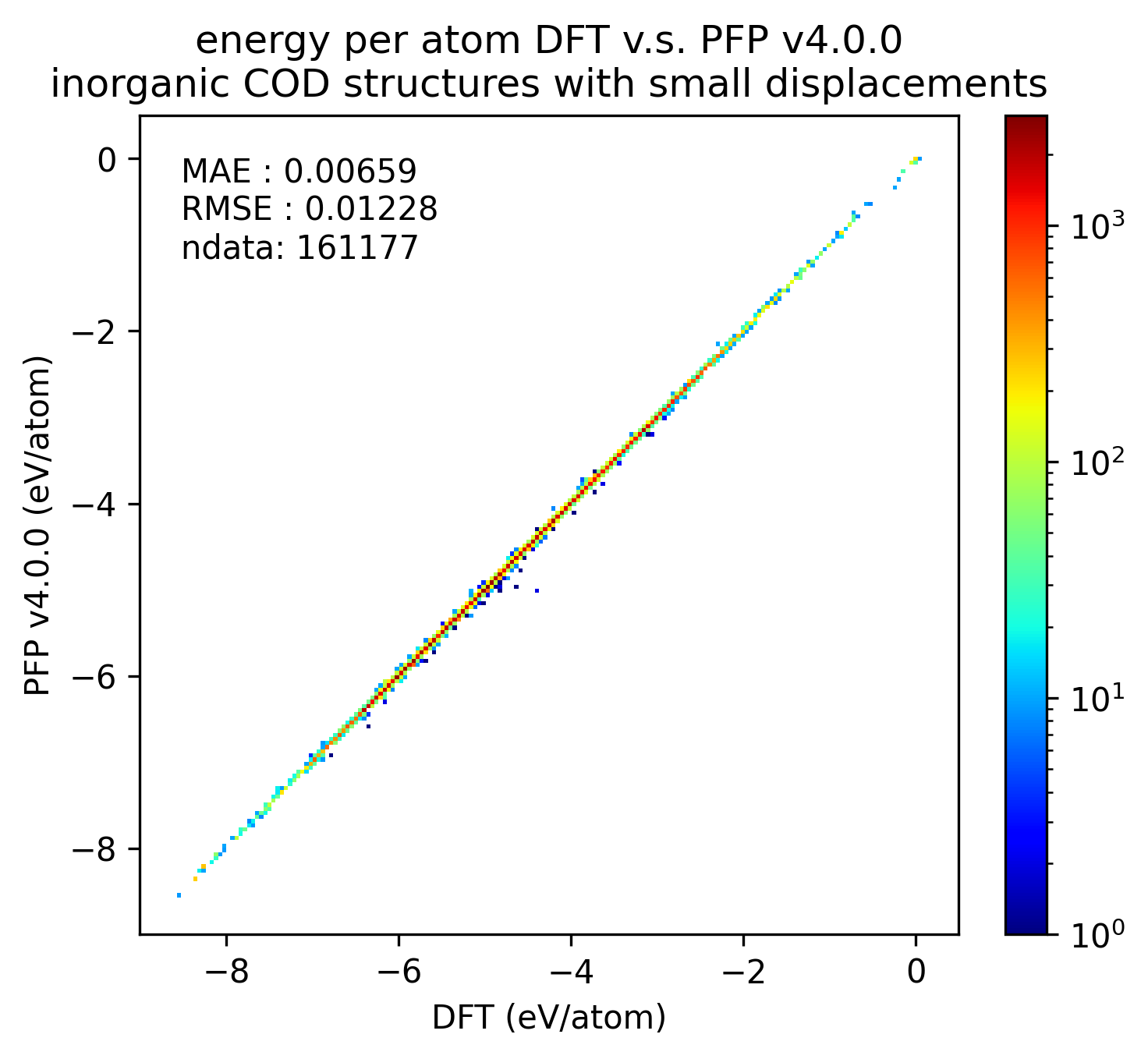 | 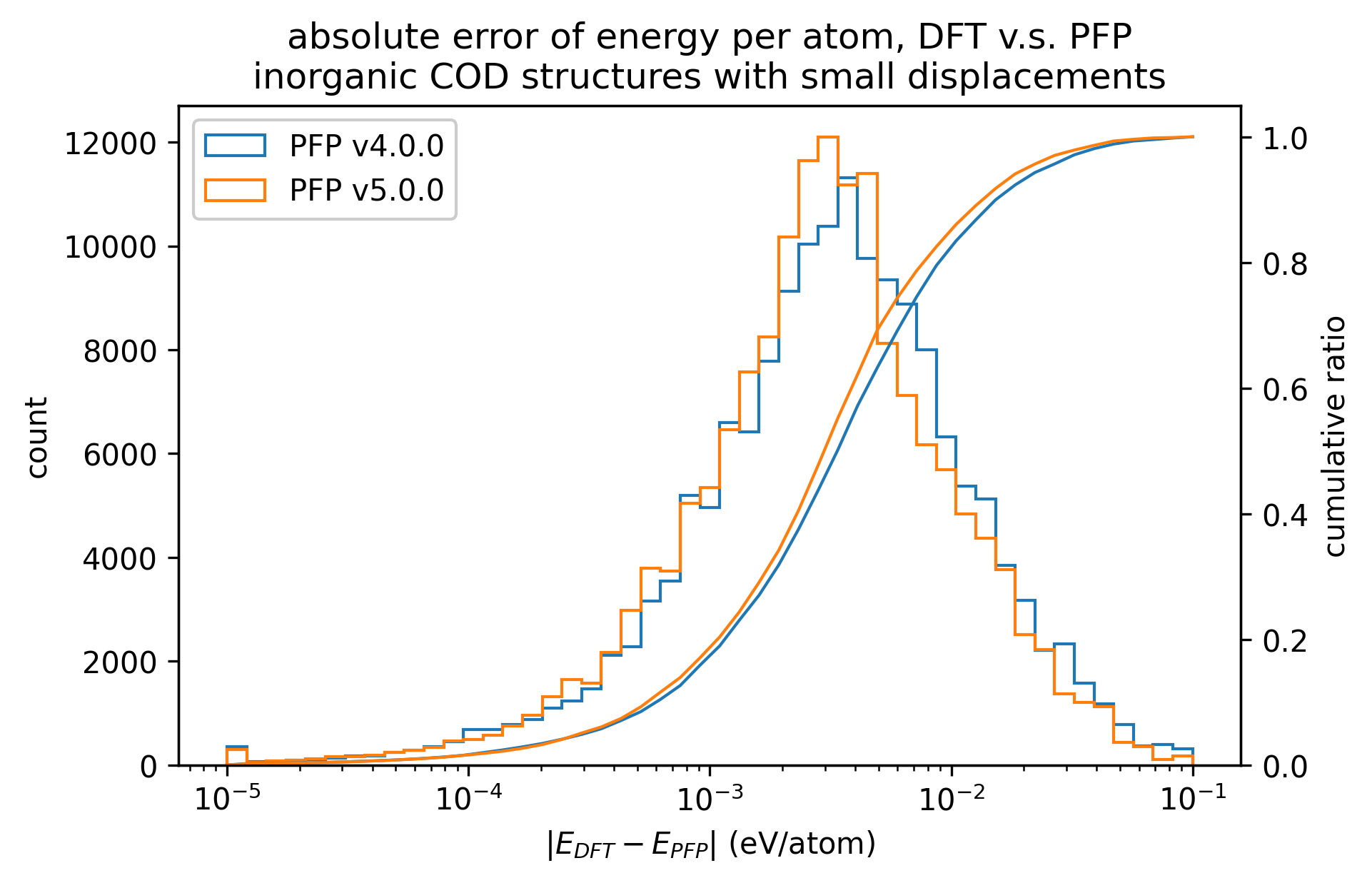 |
| 力 | 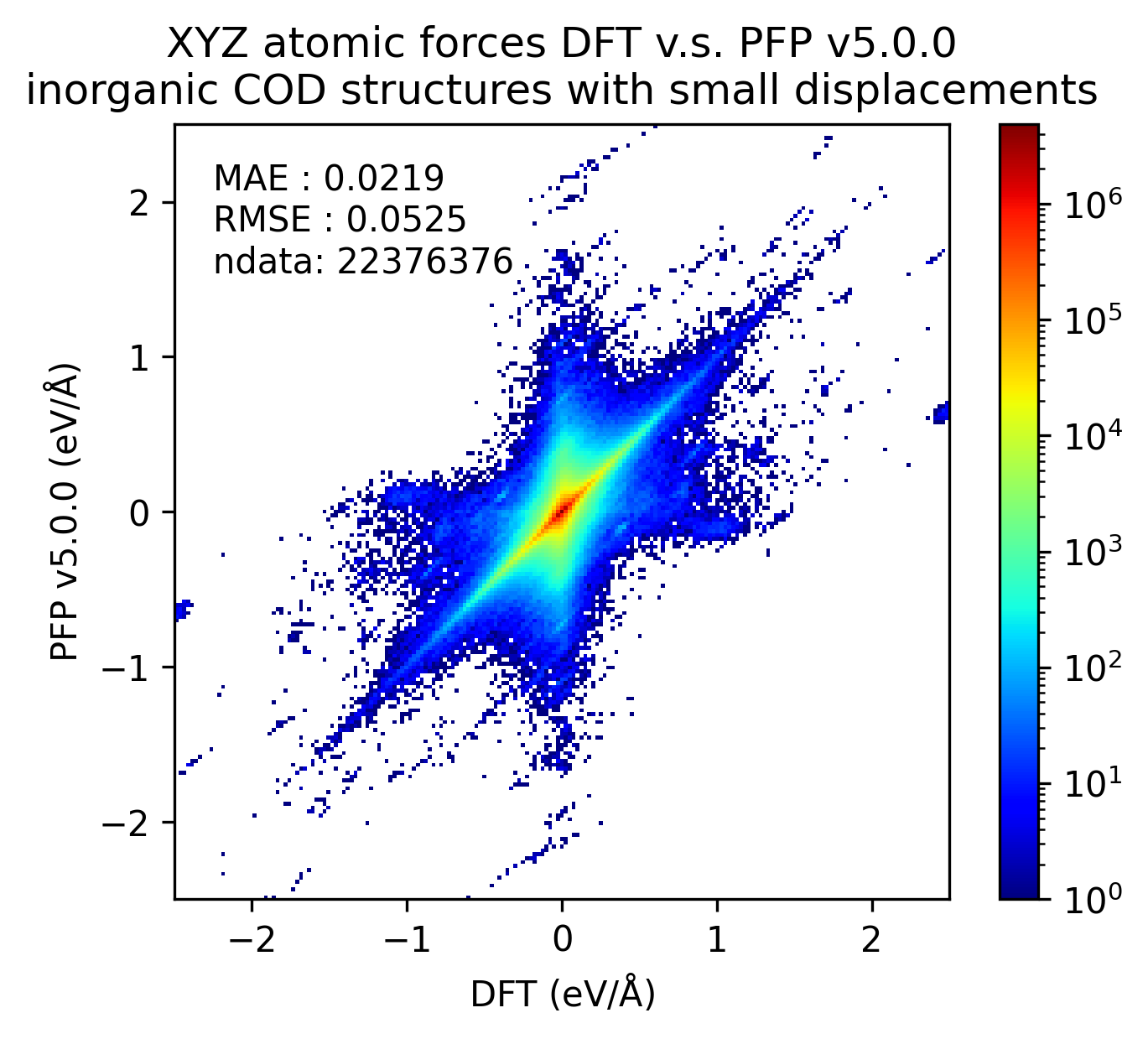 | 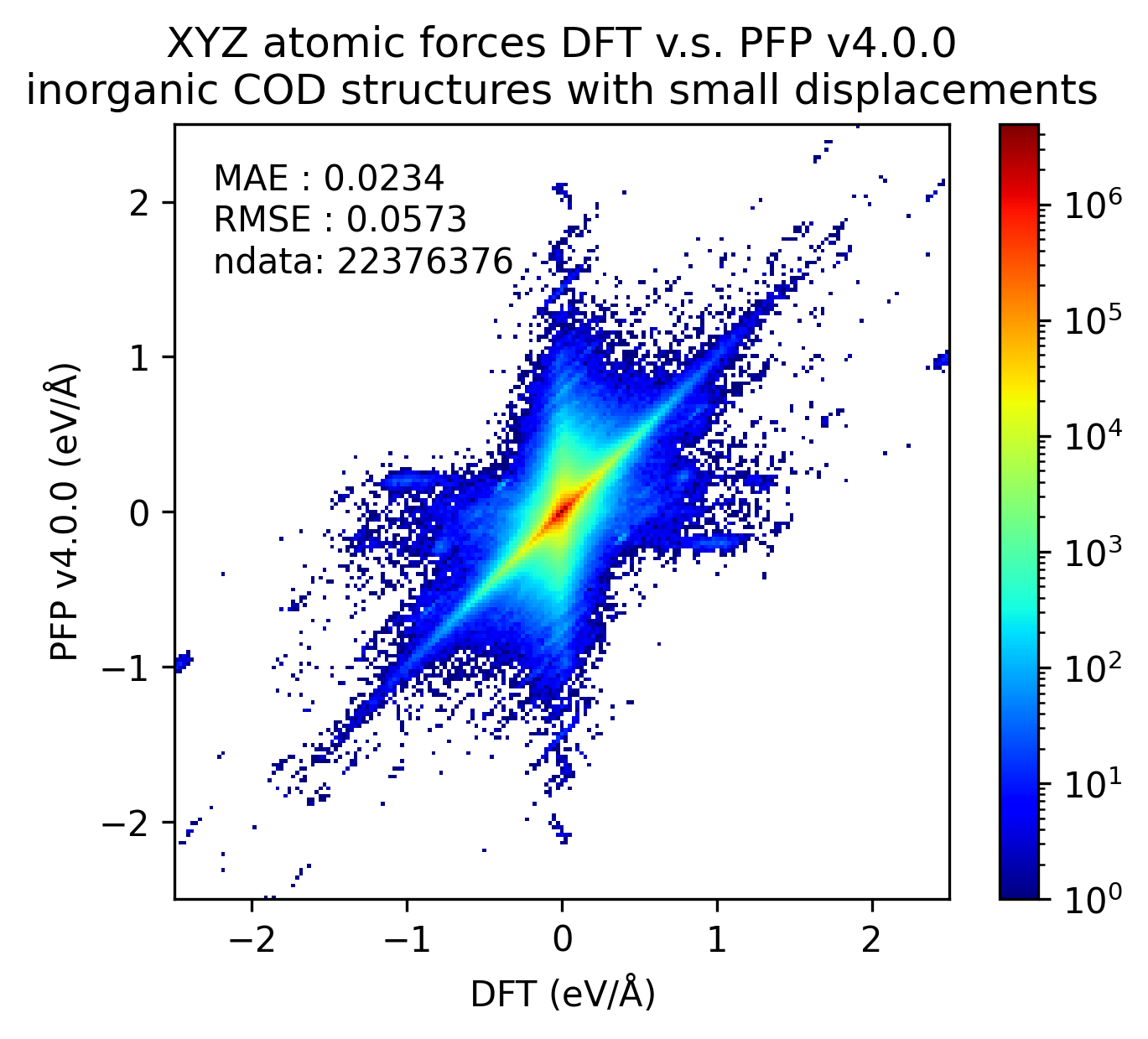 | 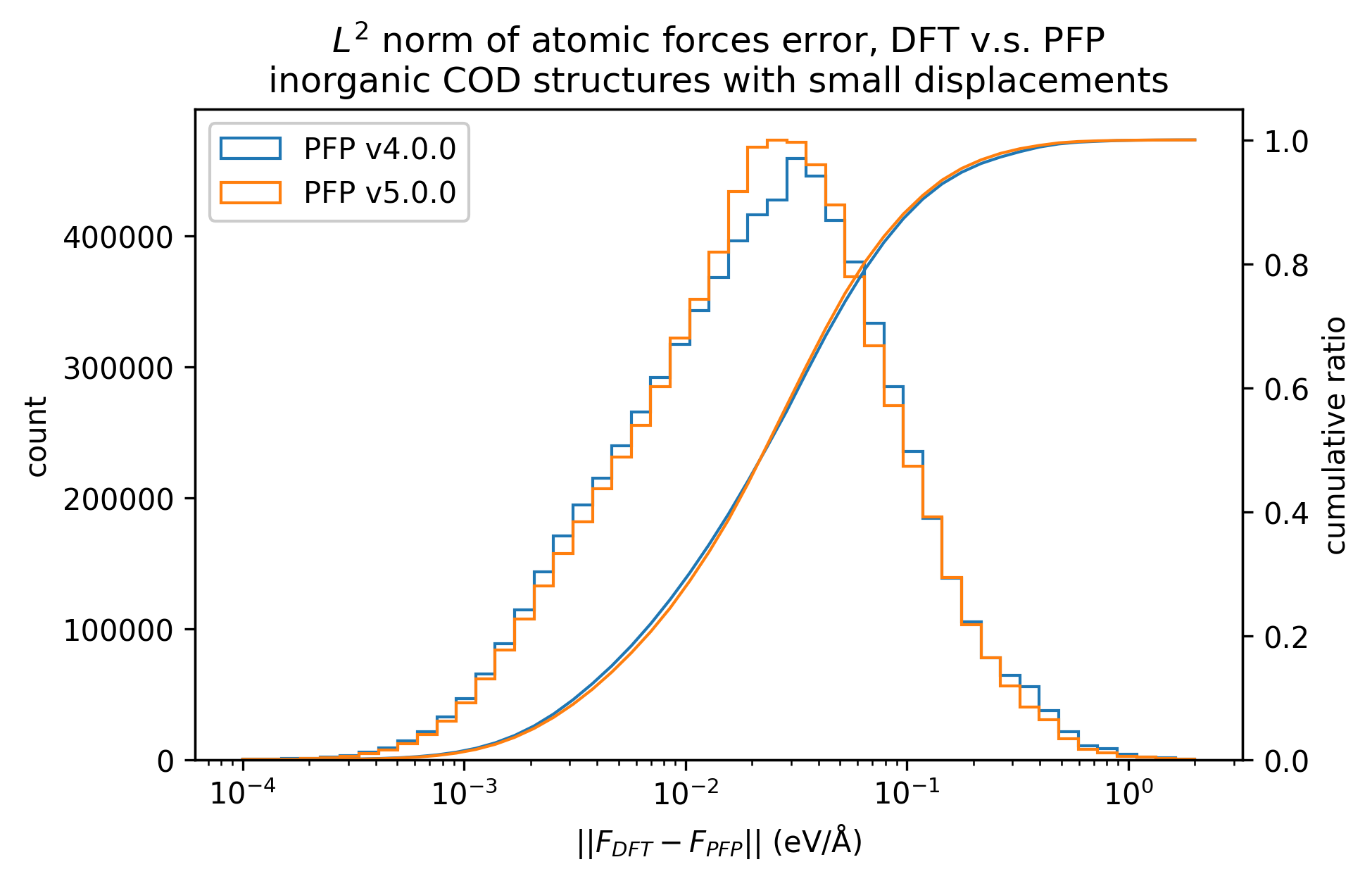 |
以上の結果から、PFP v5.0.0ではPFP v4.0.0と比較して、有機結晶と錯体結晶のエネルギーと力の再現性、すなわち、ポテンシャルエネルギー曲面の再現性が向上したと言えます。一方で、無機結晶に対してはPFP v5.0.0とv4.0.0とで同等と言えます。
Acknowledgement
PFPの最新版(v5)は、PFNのスーパーコンピュータおよび国立研究開発法人産業技術総合研究所のAI橋渡しクラウド(ABCI)を用いて開発されました。
参考資料
[1] Takamoto, So, et al. “Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements.” Nature Communications 13.1 (2022): 2991. https://www.nature.com/articles/s41467-022-30687-9
[2] “Matbench Discovery” https://matbench-discovery.materialsproject.org/
[3] “Materials Project” https://materialsproject.org/
[4] Wang, Hai-Chen, Silvana Botti, and Miguel AL Marques. “Predicting stable crystalline compounds using chemical similarity.” npj Computational Materials 7.1 (2021): 12. https://www.nature.com/articles/s41524-020-00481-6
[5] “Crystallography Open Database” https://www.crystallography.net/cod/
[6] Grimme, Stefan, et al. “A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu.” The Journal of chemical physics 132.15 (2010). https://pubs.aip.org/aip/jcp/article/132/15/154104/926936
[7] Grimme, Stefan, Stephan Ehrlich, and Lars Goerigk. “Effect of the damping function in dispersion corrected density functional theory.” Journal of computational chemistry 32.7 (2011): 1456-1465. https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.21759
-




 URLを
URLを
コピーしました
関連ニュース
-
2026.4.15プレスリリース製品・サービス
-
2026.1.28プレスリリース製品・サービス
-
2025.7.16プレスリリース企業・経営製品・サービス