Cases
導入企業の声、具体的な計算事例、論文の中から、特に注目の事例をピックアップしました。興味のあるカテゴリーから、詳細をご確認ください。
Featured Cases
Featured お客様事例
これまでの経験や勘では予測できなかった
新たな組み合わせとの出会いを創出
株式会社クラレ化学
AIや実験データと組み合わせることで、研究開発のレベルアップに計算化学が貢献。“グローバルで戦っていくための強力な武器になり得ると期待しています。”
この事例を詳しく見る
(左から) 株式会社クラレ 研究開発本部 企画管理部長 兼 デジタルソリューション部長 鎌田様、研究開発本部 本部長 須郷様、研究開発本部 デジタルソリューション部 主管 三浦様
Featured 計算事例
金属-ポリマー接合構造における界面熱抵抗計算
半導体
接着材料
界面熱抵抗
テーマ概要(背景) 界面熱抵抗は、半導体、パワーエレクトロニクス、二次電池など様々な分野で重要となる物性です。金属-金属界面、金属-ポリマー界面など材料種の組み合わせは多岐にわたりますが、特に近年は、半導体産業の後工程(パッケージング
この事例を詳しく見る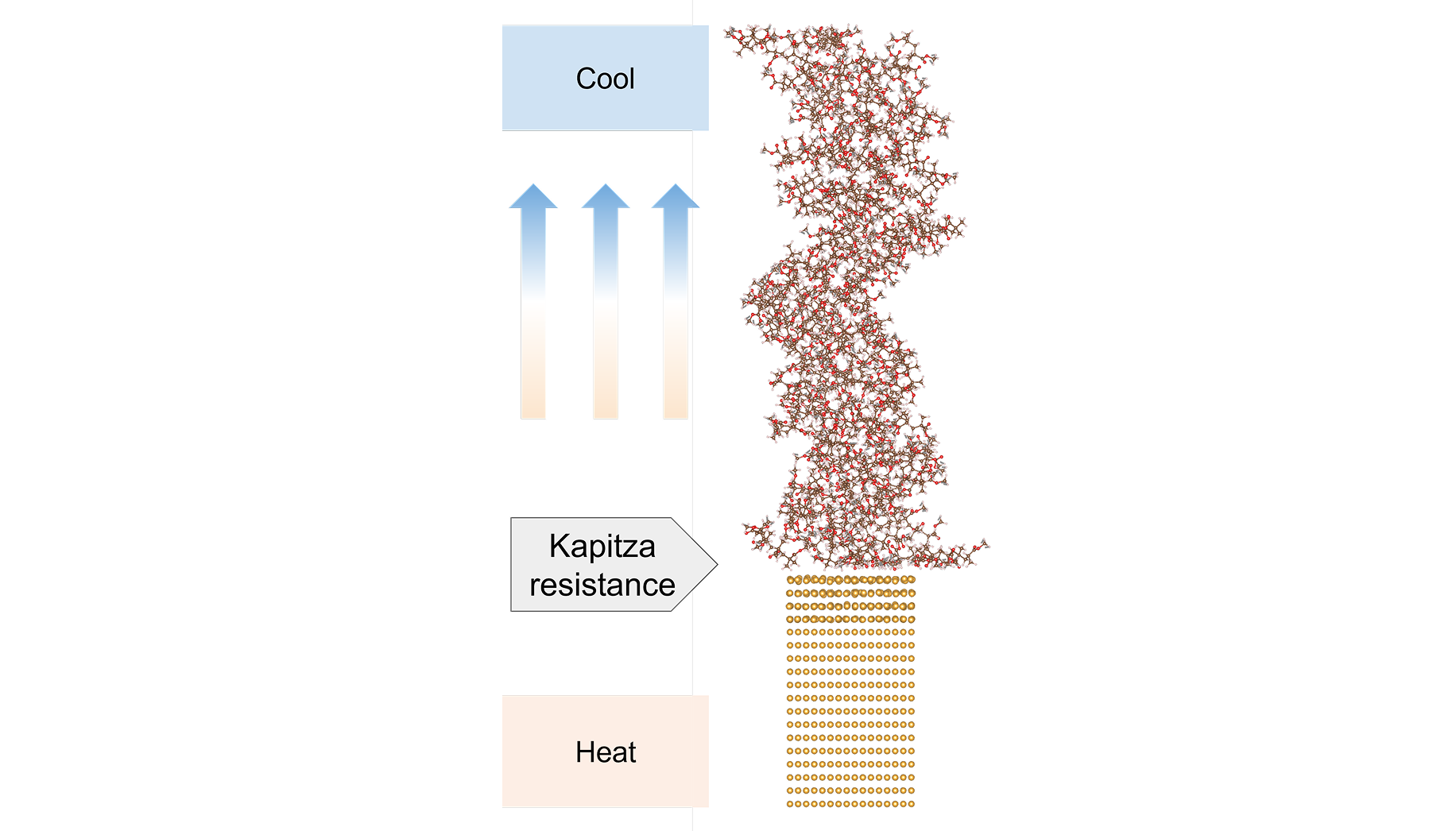
Featured 公開論文事例
Enhanced Ni Exsolution in High-Entropy Perovskite Oxides with Broadening of Migration-Reduction Energy Landscapes
Matlantisを用いた論文
セラミックス
触媒
電池
電解
While high-entropy perovskite oxides have recently emerged as promising hosts for exsolution-enabled catalysts and electrodes, a systematic understanding of how high-entropy compositions influence exsolution remains limited. Here, we compare Ni exsolution in a simpler perovskite oxide, (La0.6Sr0.4)0.95(Co0.19Fe0.76Ni0.05)O3−δ (LSCF-5Ni), and two high-entropy perovskite oxides, (La0.2Sr0.2Ca0.2Nd0.2Y0.2)0.95(Co0.19Fe0.76Ni0.05)O3−δ (CaNdY-5Ni) and (La0.2Sr0.2Ba0.2Nd0.2Y0.2)0.95(Co0.19Fe0.76Ni0.05)O3−δ (BaNdY-5Ni). The experiment reveals that the exsolved …
カテゴリから事例を探す
SNSをフォローして、事例の追加情報などをタイムリーにGET!
Matlantisの導入で、人間の直感を超える領域へ
Matlantis
ポータブルガイド
特徴から導入効果まで、この一冊でまるごと把握

-




 URLを
URLを
コピーしました