Analysis of CO2 adsorption dynamics of MOF using NEB method
Case study provided by: Institute of Science Tokyo
Theme Overview
In order to achieve a carbon-neutral society, there is an urgent need for the development of materials to capture and separate the greenhouse gas CO2. Metal–organic frameworks (MOFs), which are porous materials composed of organic multidentate ligands combined with metal ions, have attracted attention as promising candidates because of their readily modifiable structures.
In the study of reference 1, two kinds of MOFs with isolated voids which cannot be directly accessed were obtained. Despite the apparent absence of CO2 diffusion pathways in these structures, one of the MOFs was able to selectively capture and store CO2 Simulations using MatlantisTM were performed to elucidate the mechanism of this specific adsorption phenomenon.
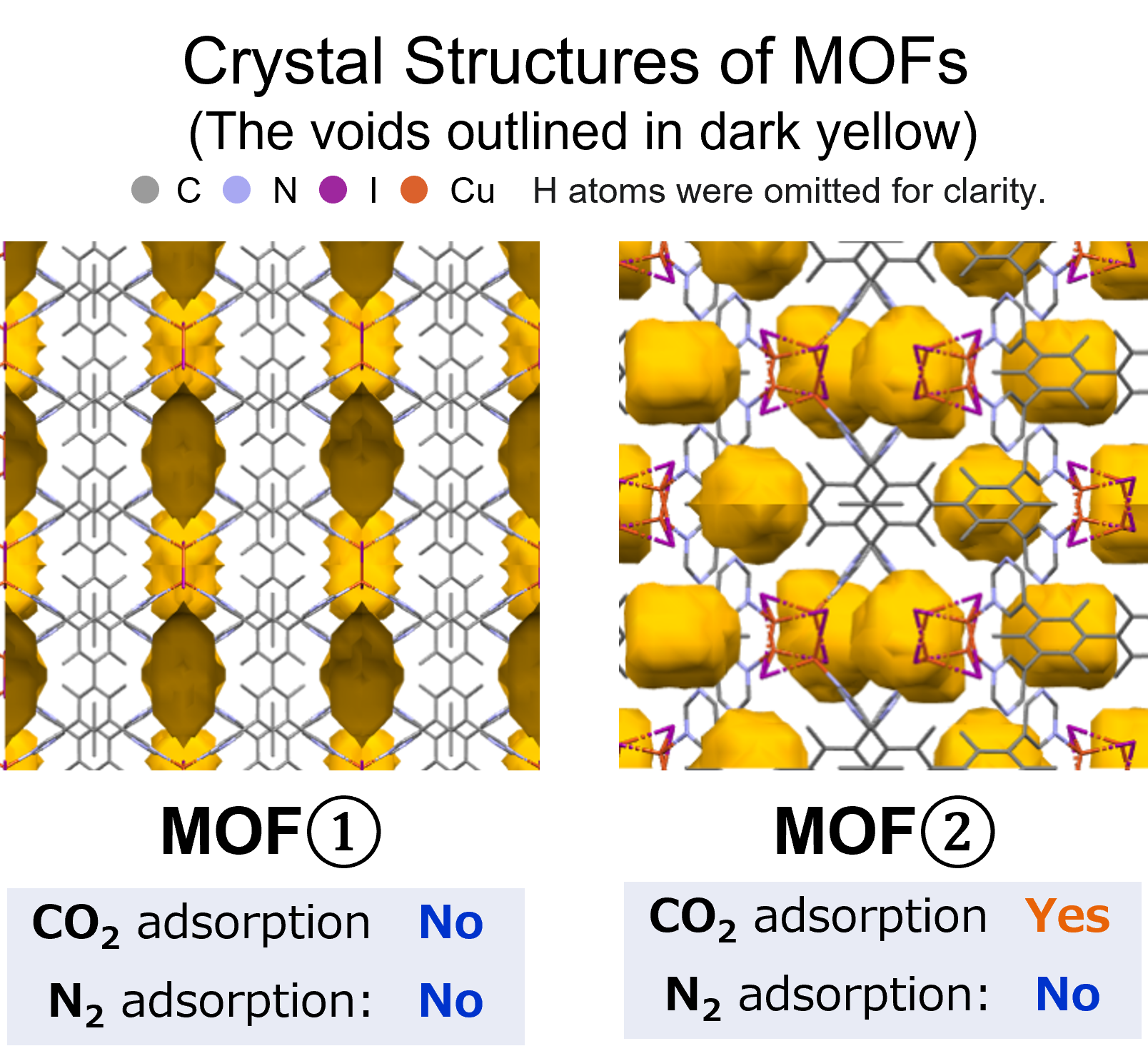
Calculation Models and Methods
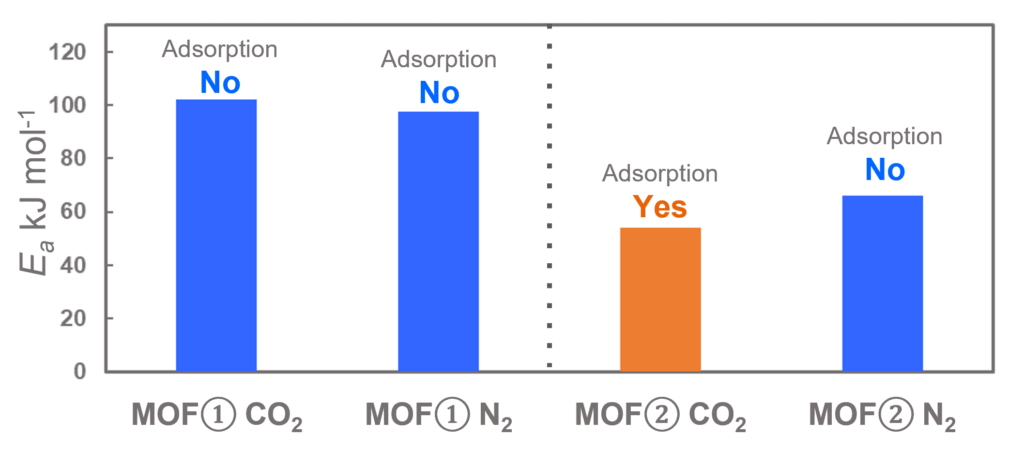
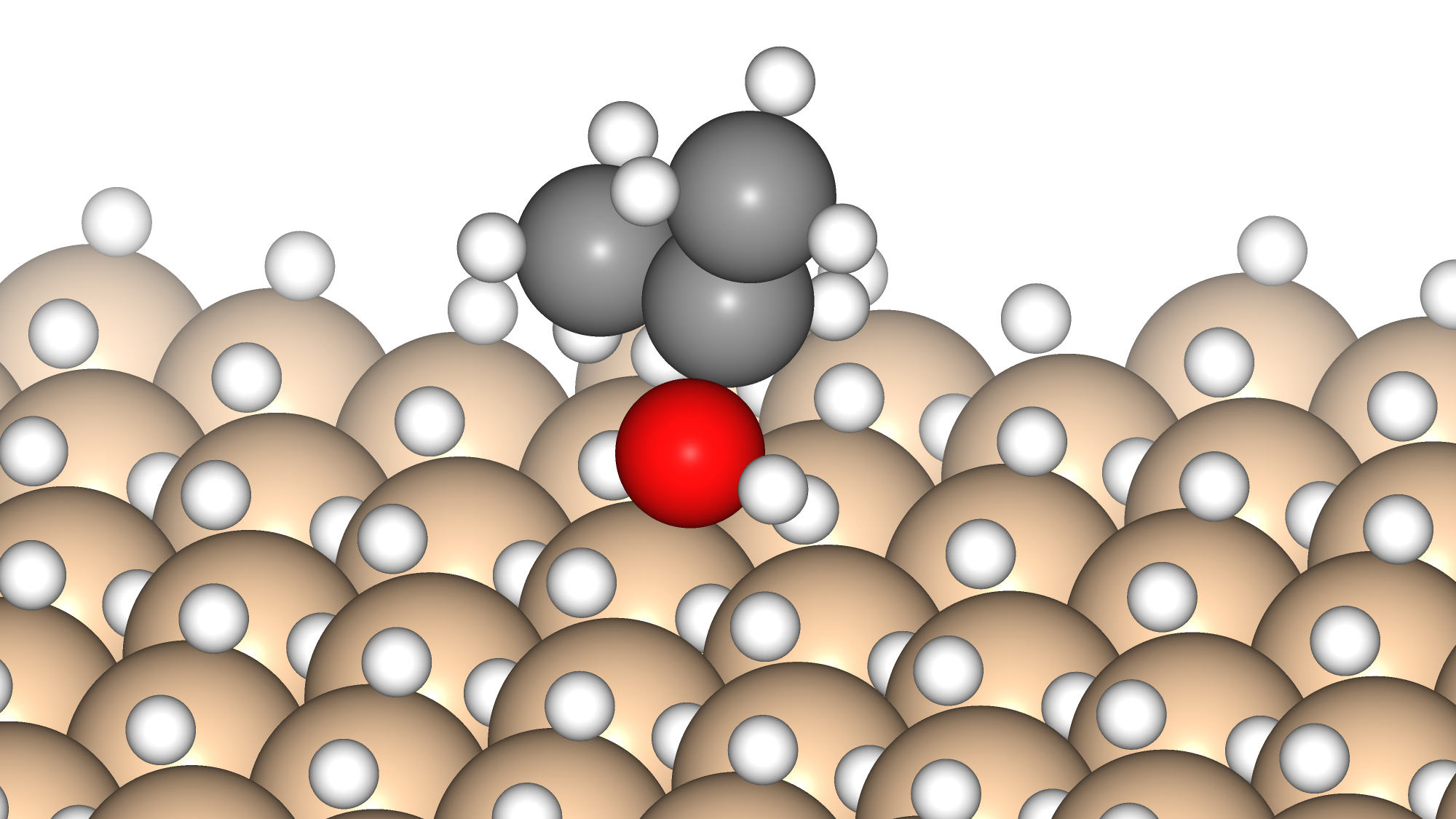
The subjects of the calculations were MOF①, which does not adsorb CO 2 or N 2, and MOF②, which does not adsorb N 2 but adsorbs CO 2. Both have pores that are isolated spaces, and at first glance, it seems impossible for molecules to enter the pores.
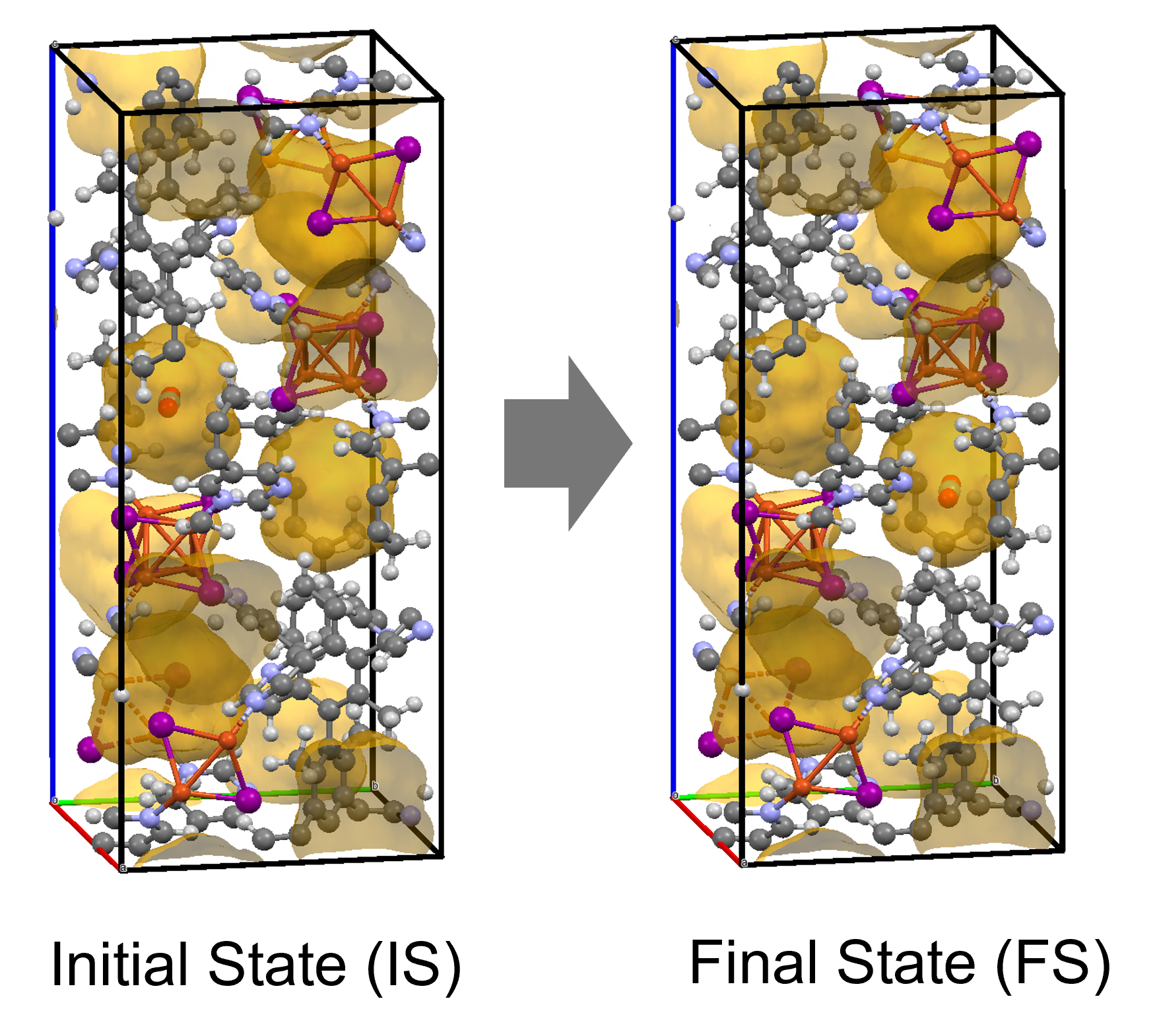
To identify the CO2 adsorption pathway, the Cl-NEB method, the method for calculating the transition state of a chemical reaction, was used. The initial state (IS) consisted of one CO2 molecule captured in one pore of the MOF, and the final state (FS) consisted of the same CO2 molecule relocated into an adjacent closed pore. By the CI-NEB method, their transition states were optimized. and the state between the initial state and the final state was calculated.
Results and Discussion
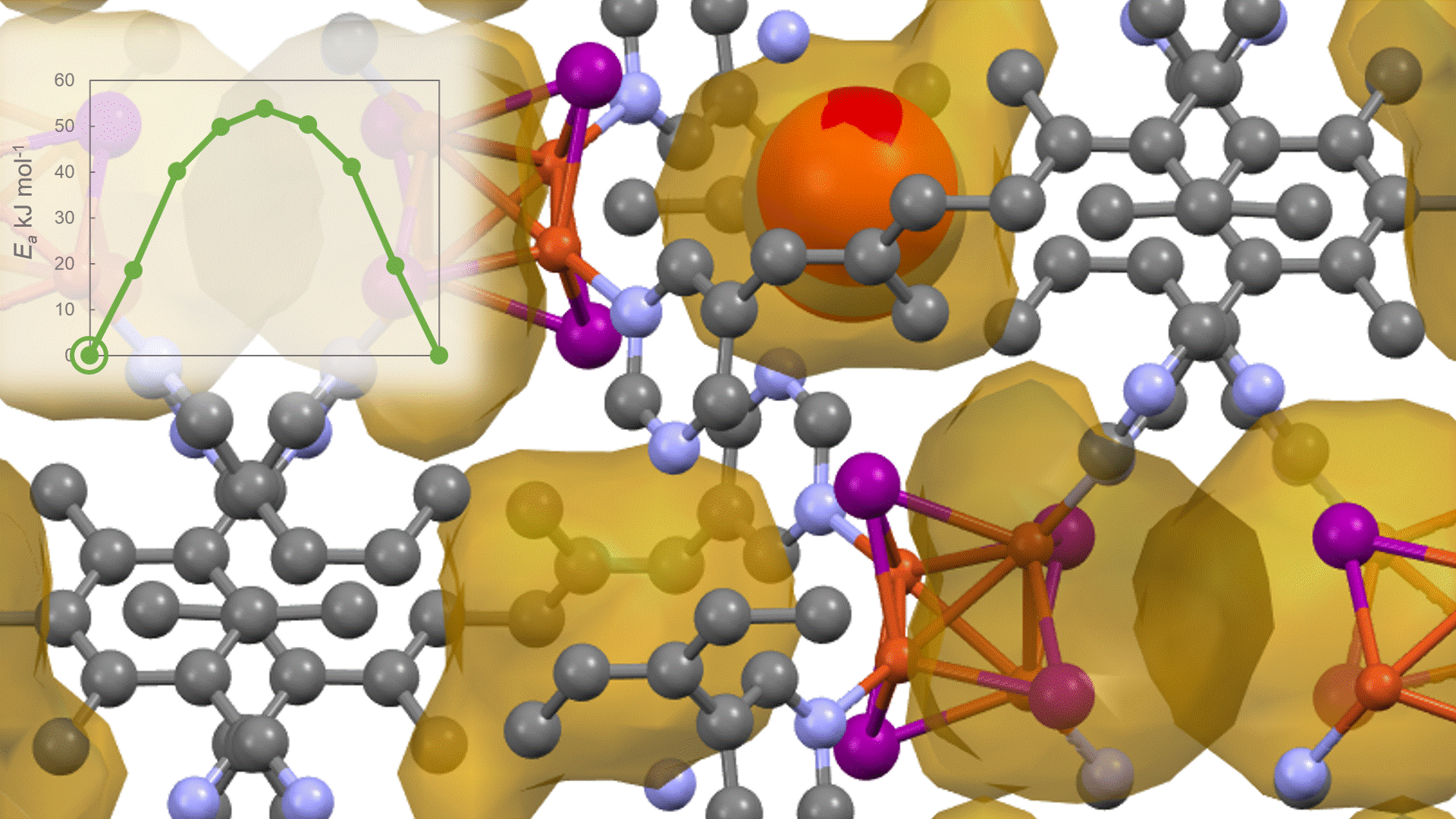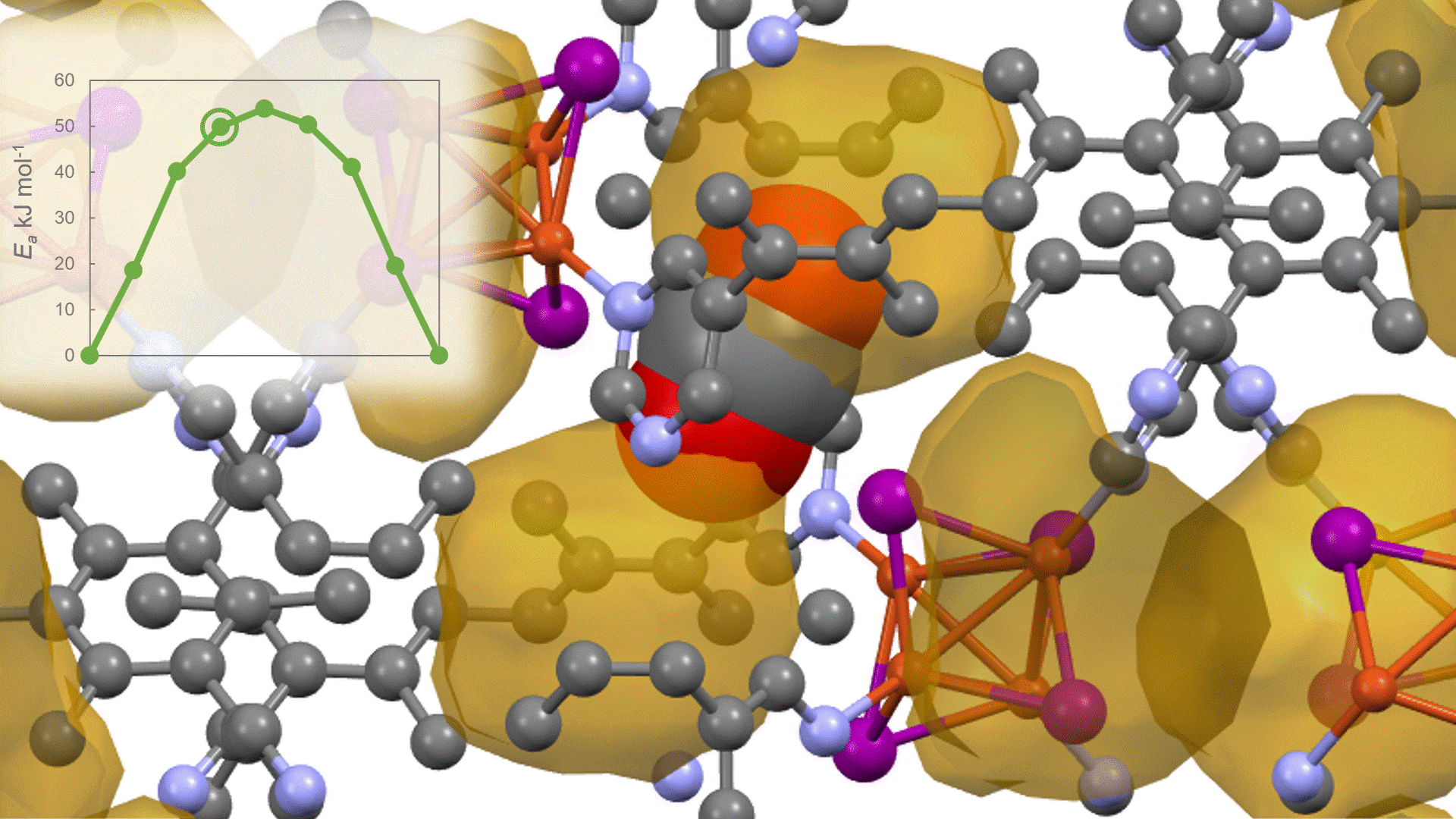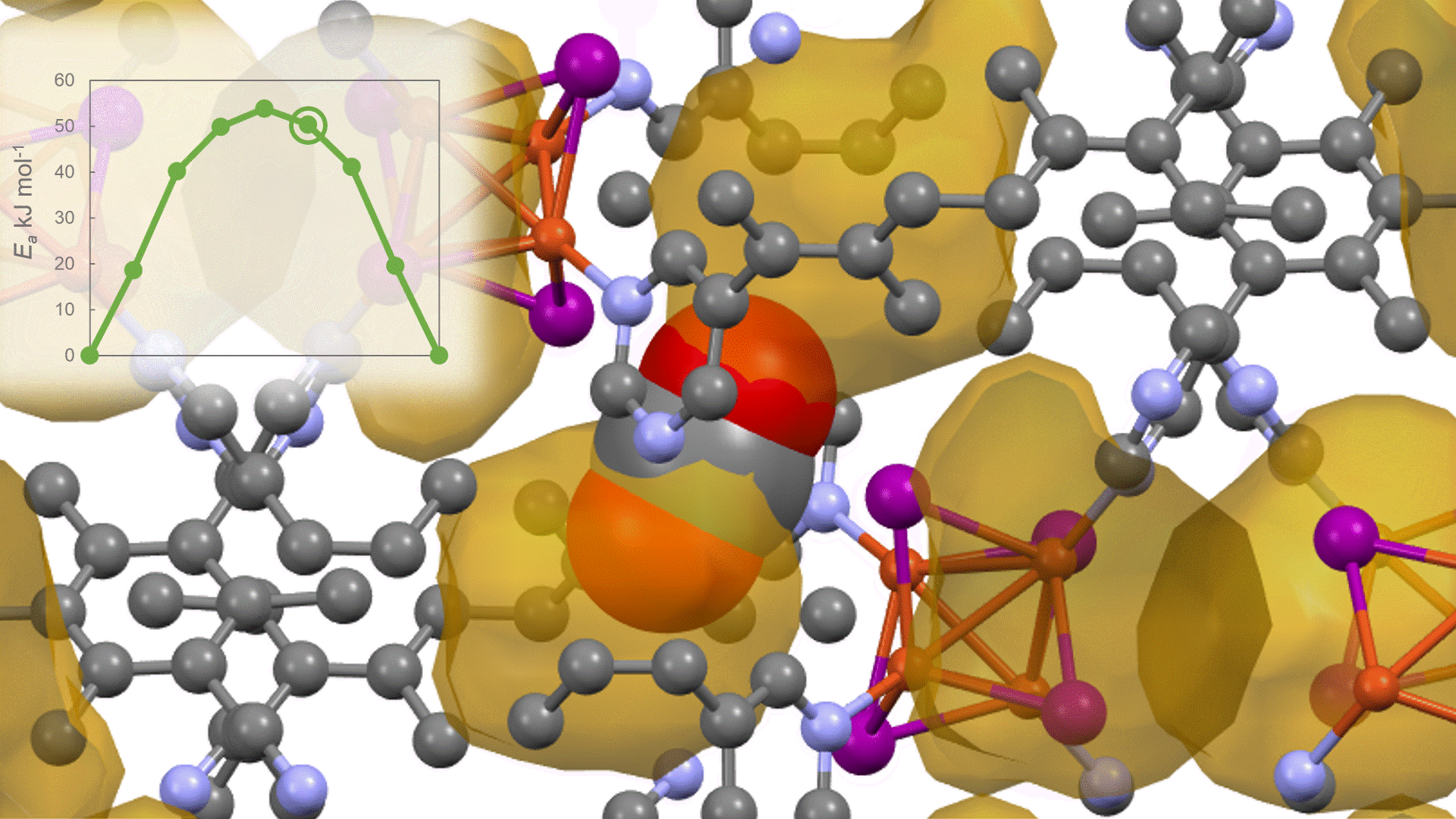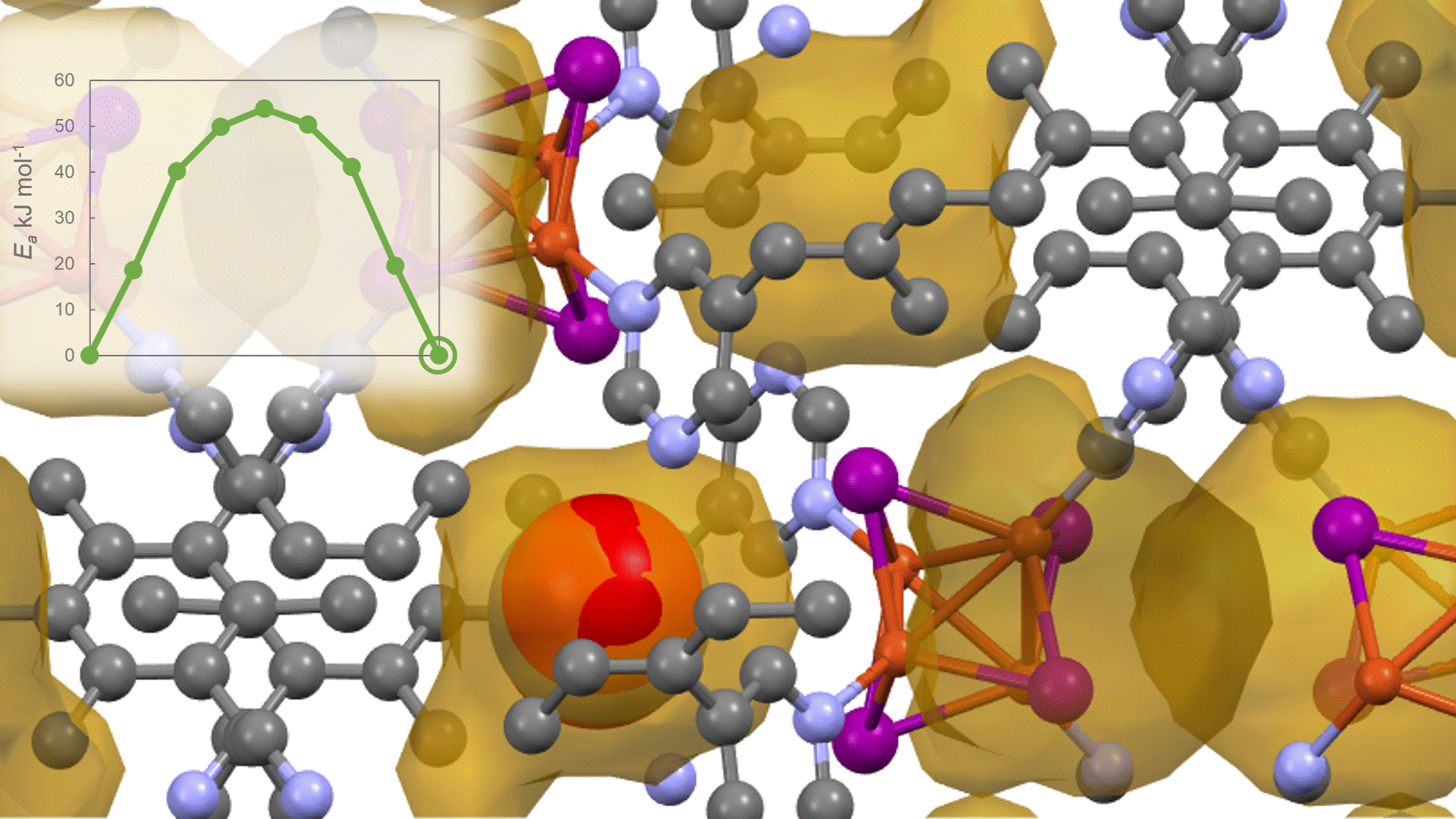
The calculation results for CO2 adsorption in MOF② are shown in the figure on the right. Only at the moment that CO2 passes through does a minute change occur in the MOF's framework structure, and a passage through which CO2 can move appears. When CO2 passes through, the passage closed behind and CO2 is trapped within the pores. This state was more energetically unstable than initial and final states, suggesting the existence of an activation barrier for CO2 adsorption.
Similar calculations were performed on the adsorption of CO 2 and N 2 in MOF ① and on the adsorption of N 2 in MOF ②. It was found that the activation barrier was larger than that for CO 2 adsorption in MOF ②, confirming the consistency with the experimentally observed results. Furthermore, X-ray crystallography did not confirm any clear structural transition of the MOF before, during, or after adsorption, and the behavior of the atoms observed in the simulation also supports the experiment.
Because of their complicates structure, the calculations of MOFs were computationally expensive and inaccurate. Thus, calculations of MOF adsorption properties have been widely used models that extract only partial structures of MOFs or models in which the atomic positions of the MOF are fixed. On the other hand, it is well known that MOFs often undergo structural changes during gas adsorption and that the flexibility of the MOF contributes greatly to the gas adsorption properties of MOFs. The ability of Matlantis to perform high-speed calculations with structural flexibility of entire MOF structures is expected to provide important insights for the design of new MOFs in the future.

Computational Details

References
[1] T. Shimada et al. Adv. Sci. 2024, 11, 2307417 [2] G. Henkelman et al. J. Chem. Phys. 2000, 113, 9901.Related Documents
[3] Lecture video [4] Tokyo Institute of Technology websiteSelective capture of CO2 by a novel "Magic door" mechanism: Creation of innovative MOF materials toward the realization of a carbon-neutral society | Tokyo Tech News | Tokyo Institute of Technology (titech.ac.jp) [5] ENEOS Corporation website
Research results on selective capture of CO2 using a novel "Magic door" mechanism published in Advanced Science (November 2023) | Laboratory NEWS & TOPICS | Research and Development Special Site (eneos-rd.com)
Profile of the case study provider
Institute of Science Tokyo

tag
Publication date of this case: 2024.07.11
-




 URL
URL
Copied
what's new
High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
Pharmaceuticals
Data-Driven Method for Discovering Low Adsorption Energy Molecules on Si Surfaces
ScreeningMaterials InformaticsSemiconductors
Calculation and Verification of Methanol Synthesis Catalysts
Methanol synthesis catalystsLarge-scale screening
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis
