Calculation of the activation barrier for CO dissociation on Cobalt Fischer-Tropsch Catalysts
Renewable Synthetic Fuel
This is an example of a catalyst for "renewable synthetic fuel" using hydrogen derived from renewable energy and CO2 as feedstocks.
The reaction that produces hydrocarbons, which are synthetic fuel, is called the Fischer-Tropsch (FT) reaction, and hydrocarbons are produced from "hydrogen" and "CO" using a catalyst. Cobalt or iron is used as the catalyst.
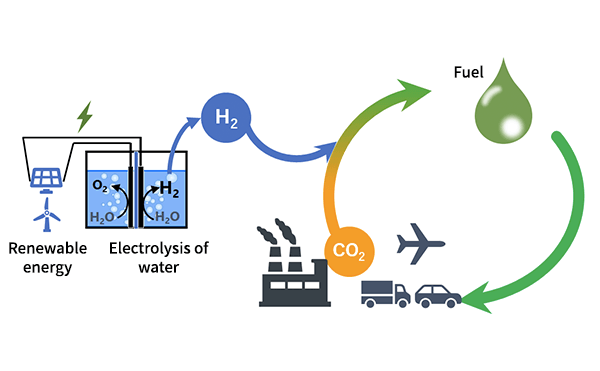
Fischer-Tropsch (FT) Reaction
The FT reaction is a complex reaction consisting of many elementary reactions, among which the CO dissociation reaction is considered to be one of the rate-determining reactions [1][2]. Here, the activation energy of CO dissociation in a cobalt catalyst was calculated by Matlantis.
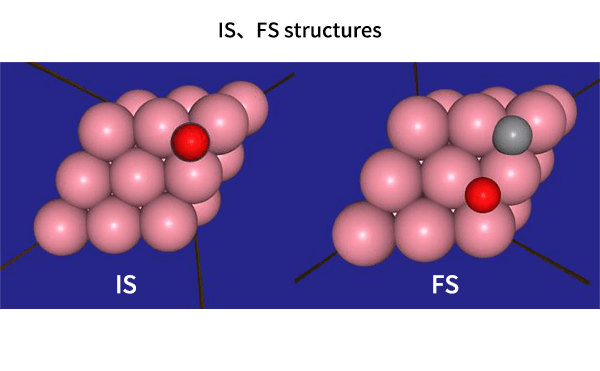
○: Hydrogen ●: Oxygen ●: Carbon ●: Co catalyst

Calculation Models and Methods
Here, we use the Nudged Elastic Band (NEB) method to calculate the activation energy of the CO dissociation reaction on the Co(0001) surface. The base model of the bulk crystalline cobalt structure is obtained from the Materials Project database.
The obtained structure is used to make a model for NEB calculations. Once the Co(0001) surface is made, the initial state (IS) and final state (FS) are constructed with adsorbed CO molecules.
Using IS and FS structures, the NEB calculation was performed.
Results and Discussion
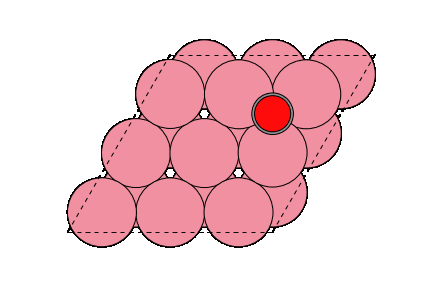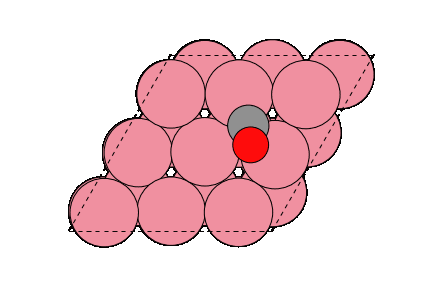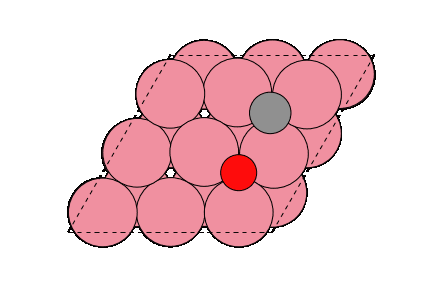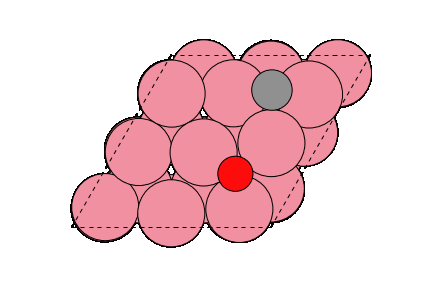
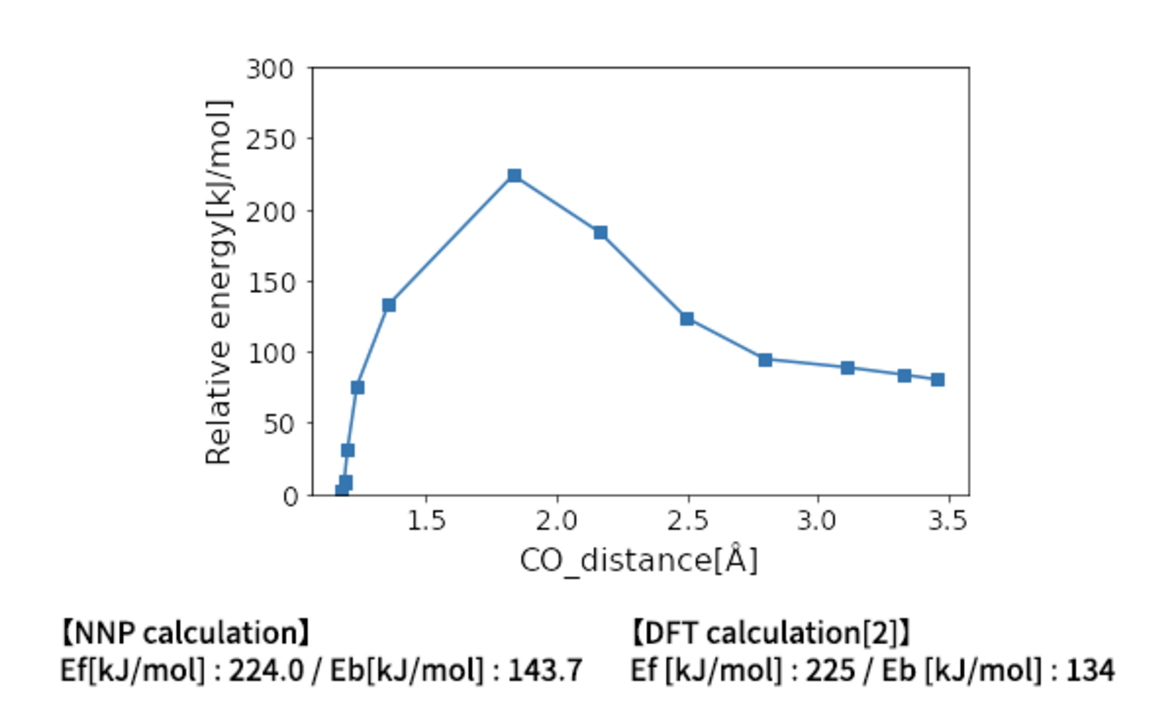
Matlantis is able to calculate the activation energy of the CO dissociation with the same accuracy as in the previous literature [2]. A conventional NEB calculation using DFT is often computationally very expensive, but it can be expedited by PFP.
PFP has learned many elements and can perform thousands of activation energy calculations in a reasonable amount of time. Therefore, it is possible to explore a wide range of catalysts with high performance.
For example, vanadium(V) promoted cobalt surface created by substituting a V atom for a Co atom can promote the CO dissociation reaction. In fact, it is known experimentally that vanadium can promote FT reactions [5, 6].
Because of the universality and blazing speed of PFP, it can not only elucidate the adsorption, physical properties, and phenomena of a single composition, but also screen materials that composed of multiple elements and structures.
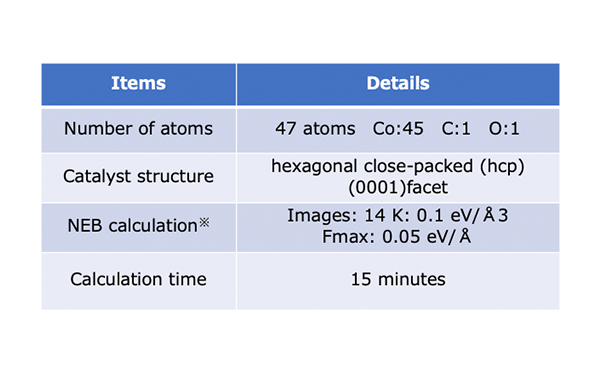
Computational Details
References
[1] Bart Zijlstra et. at., ACS Catal., 10, 9376-9400, (2020) https://pubs.acs.org/doi/full/10.1021/acscatal.0c02420 [2] Bart Zijlstra et. at., ACS Catal., 9, 7365-7372, (2019) https://pubs.acs.org/doi/full/10.1021/acscatal.9b01967 [3] https://www.materialsproject.org/materials/mp-54/ [4] https://wiki.fysik.dtu.dk/ase/ase/neb.html [5] T. Wang et. al., Catal. Lett., 107, 47 (2006) https://link.springer.com/article/10.1007/s10562-005-9730-1 [6] K. Shimura et.al, Appl. Catal. A, 494, 1 (2015) https://www.sciencedirect.com/science/article/abs/pii/S0926860X15000320tag
Published: July 1, 2021
-




 URL
URL
Copied
what's new
High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
Bio and Pharma
Data-Driven Method for Discovering Low Adsorption Energy Molecules on Si Surfaces
ScreeningMaterials InformaticsSemiconductors
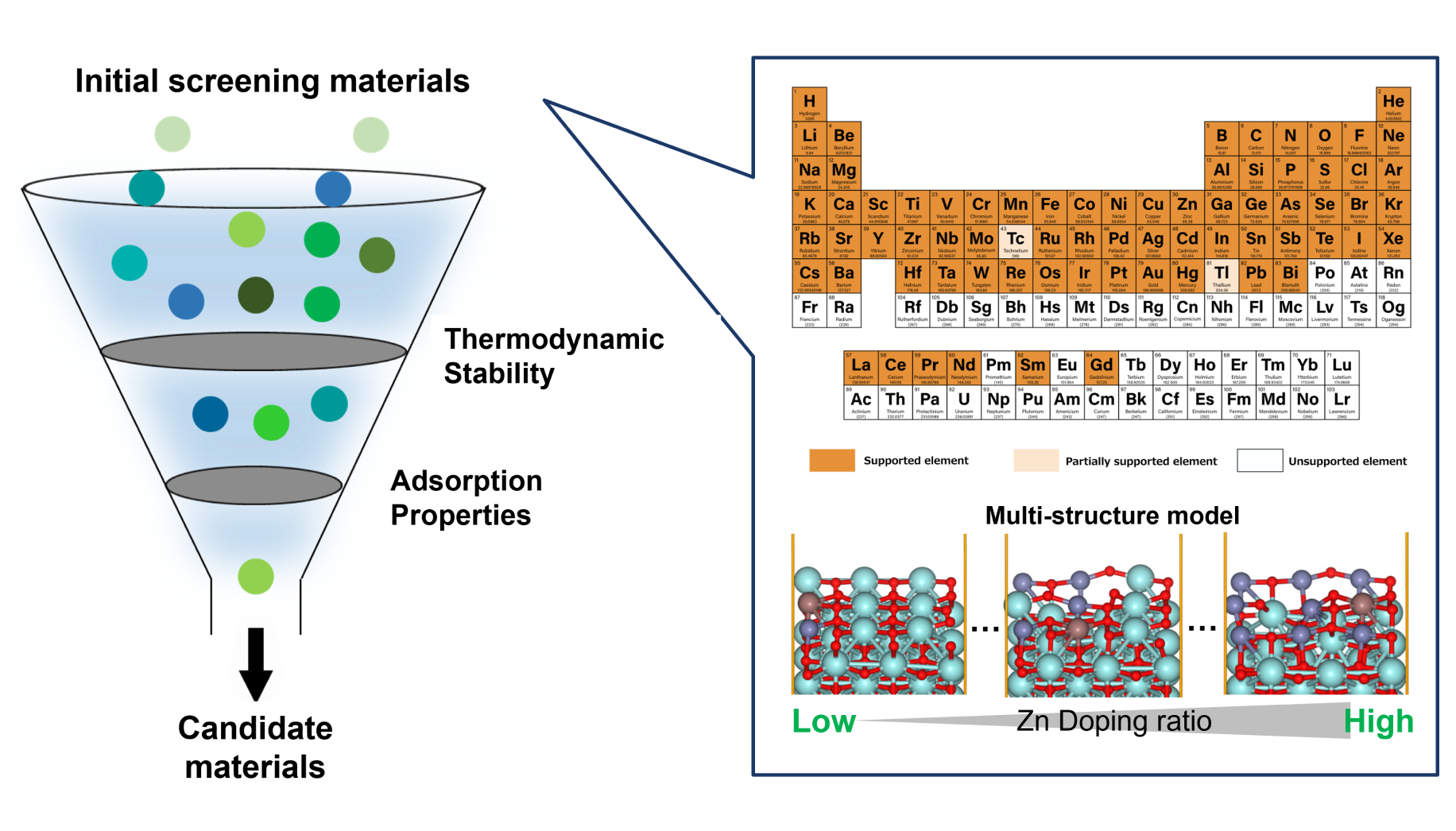
Calculation and Verification of Methanol Synthesis Catalysts
Methanol synthesis catalystsLarge-scale screening
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis
