Catalyst Screening for Ammonia Synthesis Catalyst
Case study provided by: ENEOS Corporation
Theme Overview
Heterogeneous catalysts play an important role in the production and utilization of fuels, chemicals, and pollution remediation. Virtual screening has become a popular method for finding better catalysts. One of the most successful methods for screening heterogeneous catalysts is the descriptor-based microkinetics model based on scaling relationships [1]. In this method, catalytic activity is plotted as a function of one or two descriptor energies, resulting in a volcano-shaped plot. The catalytic activity exhibits a volcano-shaped shape, called a volcano plot. This plot can provide guidance on the optimal catalyst to maximize catalytic activity and selectivity.
In this study, we attempted fast catalyst screening by using Matlantis to calculate the descriptor energy using DFT, which has a high computational cost. As a model catalytic reaction, we selected ammonia synthesis, one of the most important reactions in industry. Using Matlantis, the reproduction of DFT calculation results, as well as the virtual screening of new candidate catalysts, were examined.
Calculation Models and Methods
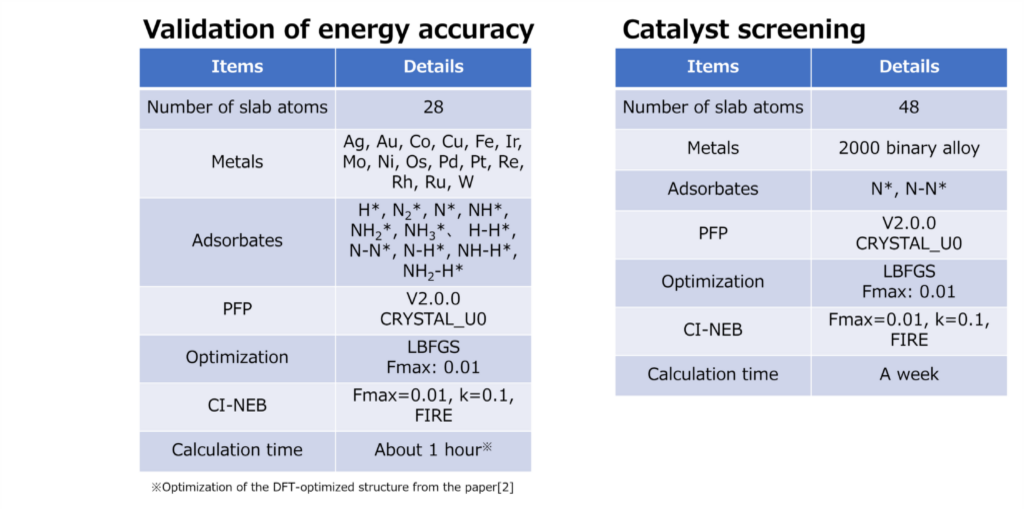
Validation of energy accuracy
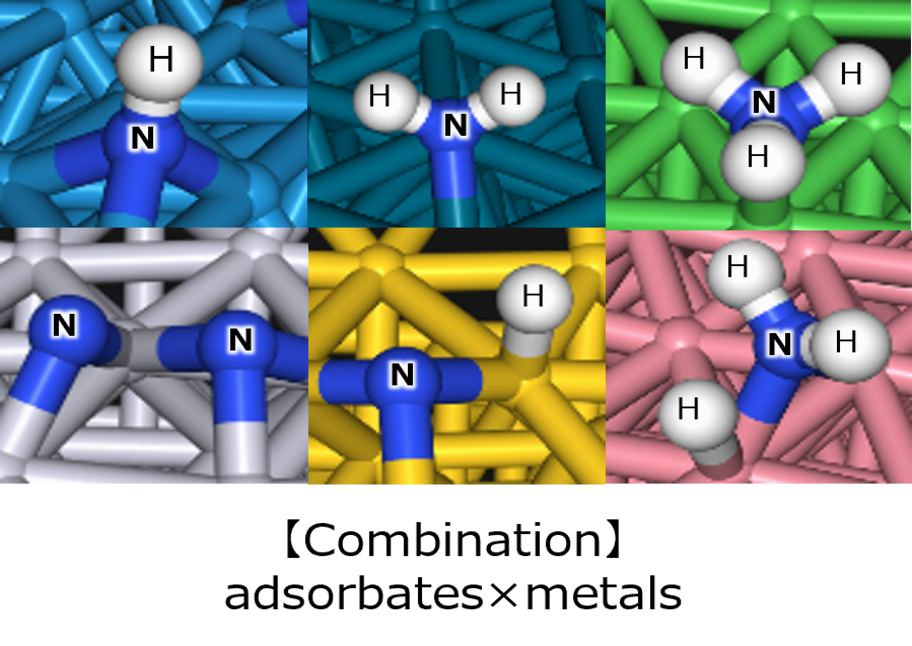
Energy calculations were performed using Matlantis for intermediates (slab, H*, N 2*, N*, NH*, NH 2*, NH 3*), transition state (HH*, NN*, NH*, NH-H*, NH 2-H*) related to ammonia synthesis adsorbed on various transition metals. A comparison was made between the PFP and DFT results [2].
(The asterisk(*) symbol indicates an adsorbed molecule; for example, N* indicates that an adsorbed N atom is adsorbed on the catalyst surface.)
Descriptor-based microkinetics for ammonia synthesis
Using the energies calculated by Matlantis, descriptor-based microkinetic modeling was performed to simulate the ammonia synthesis reaction rate. The calculation tool CatMAP [3] was used. The ammonia synthesis rate was calculated under the conditions of 673K, 100 bar total pressure, H 2: N 2 = 3:1, and 2% NH 3 conversion.
Catalyst Screening
Calculations of the N* adsorption energy (EN *) and NN* transition state energy (E N-N *) were performed on approximately 2,000 different binary alloy catalysts [4]. Finally, the calculated energy results were plotted on a map constructed from the ammonia synthesis rate calculations, enabling virtual screening of new catalysts.
Results and Discussion
Validation of energy acuracy
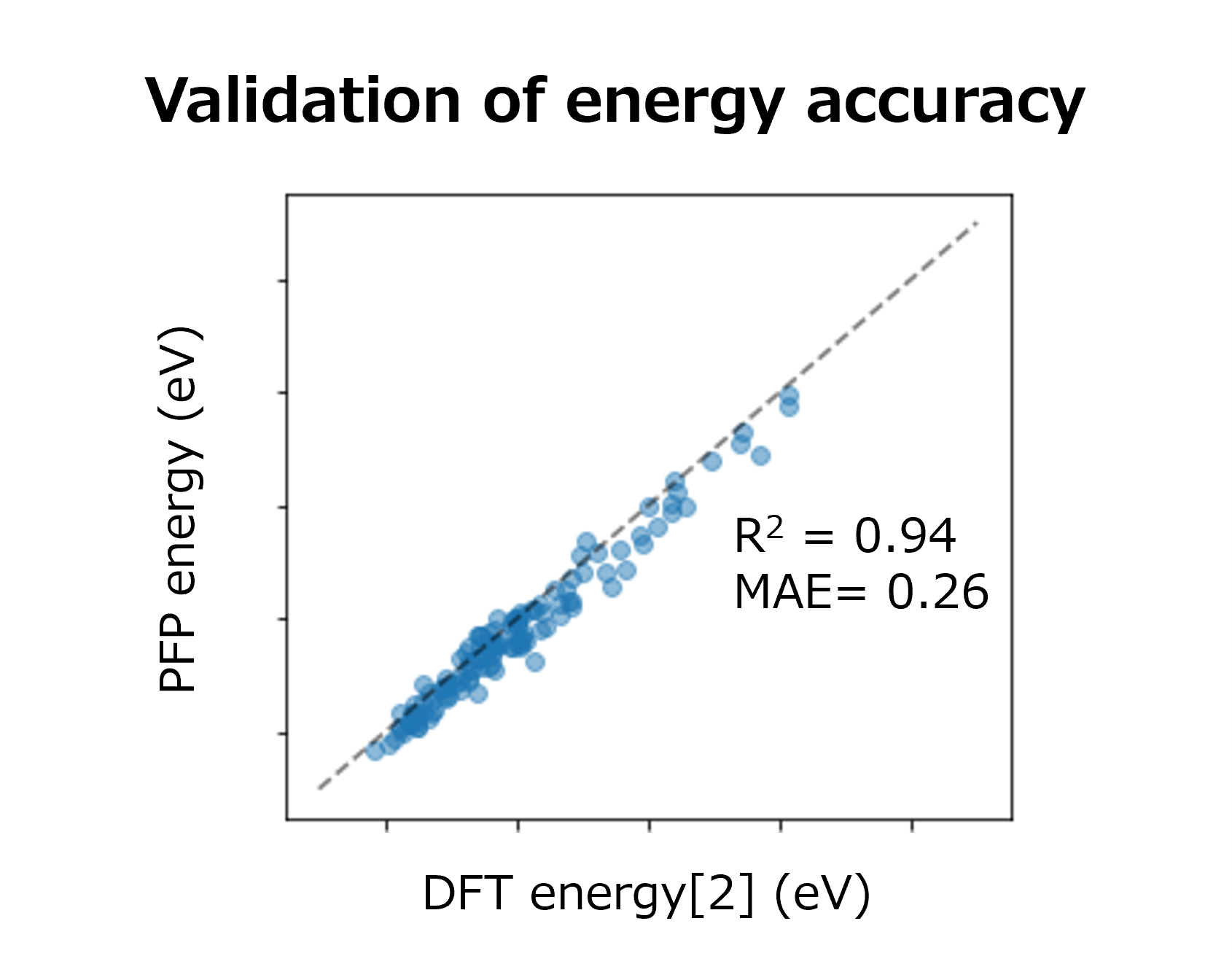
The results of a comparison between the energies calculated by PFP and DFT [2] are shown in the figure. Each energy value is plotted linearly, demonstrating that PFP calculation can be performed with the same accuracy as DFT (R2=0.94) regardless of the type of metal or surface chemical species. This indicates that Matlantis possesses both high universality and accuracy.
Descriptor-based microkinetics for ammonia synthesis
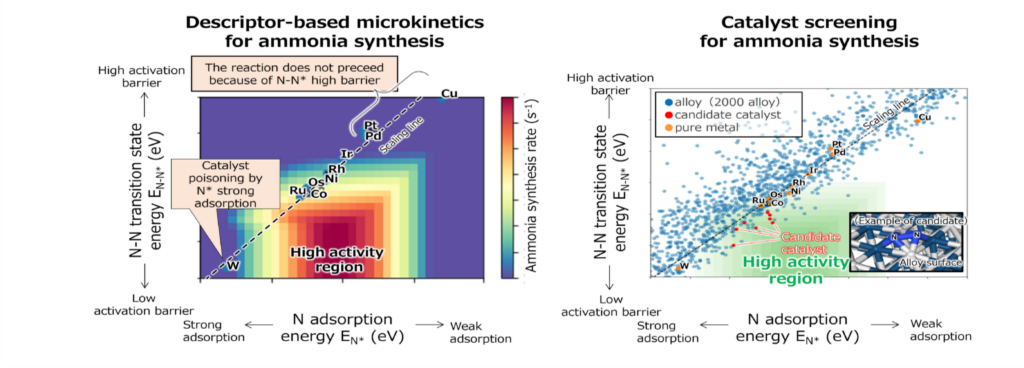
The x axis of the figure represents the adsorption energy of N* (EN*), and the y axis represents the transition state energy of the N-N* (EN-N*). The plot shows EN * and E N-N* values for each transition metal calculated by PFP. This study, which combines Matlantis with descriptor-based microkinetics, calculated figures with accuracy comparable to previous DFT work [2].
Catalyst Screening
Taking advantage of Matlantis's high speed, high accuracy, and universality, EN* and EN-N* calculations were performed on approximately 2,000 different binary alloy catalysts, enabling the virtual screening of new catalysts. As a result, several promising candidate catalysts (red plots) were identified, which are predicted to have high activity for ammonia synthesis.
Although Catalyst screening using descriptor-based Microkinetics has been constructed in previous studies, it takes a long time to calculate, especially when transition states are calculated to determine required descriptors. In this study, multiple promising candidate catalysts were identified in a short period. It is expected that this method will become a powerful tool for discovering new catalysts in the future.
Computational Details
References
[1] Yue Xu et. al., New J. Phys., 15 12521, (2013) [2] Tao Wang et. al., PNAS, 118, (2021) [3] A.J. Medford et. al., Catalysis Letters, 145, 3, 794-807(2015) [4] Osman Mamun et. al., SCIENTIFIC DATA, 6:76, (2019)Profile of the case study provider
ENEOS Corporation

tag
Publication date of this case: 2024.11.13
-




 URL
URL
Copied
what's new
NEW
High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
Pharmaceuticals
Data-Driven Method for Discovering Low Adsorption Energy Molecules on Si Surfaces
ScreeningMaterials InformaticsSemiconductors
Calculation and Verification of Methanol Synthesis Catalysts
Methanol synthesis catalystsLarge-scale screening
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis
