Evaluation of Lithium Ion Conductivity of Garnet Type Oxide Materials
Case study provided by: Nagoya Institute of Technology Nakayama lab.
Theme Overview
Current lithium-ion batteries use flammable organic electrolytes, which poses the risk of fire and explosion. Safe batteries can be created by using non-flammable lithium-ion conductive oxides as solid electrolytes, but it is necessary to find materials with sufficiently high lithium-ion conductivity. First-principles molecular dynamics (FPMD) is a promising material search approach that can replace experimental trial-and-error methods because it can evaluate ionic conductivity with high accuracy. However, even with current computers, the calculation costs are high, and it often takes several weeks to several months to evaluate a single composition and a single condition (temperature, etc.).
In this study, we performed molecular dynamics (MD) simulations using the fast and accurate Matlantis software on a garnet-type Li7La3Zr2O12 material, a typical oxide solid electrolyte material, to evaluate the Li ion conductivity. We also compared the results with those of FPMD calculations performed under the same conditions.
Calculation Models and Methods
A model structure of Li56La24Zr16O96 (total 192 atoms) was constructed, and constant-volume first-principles molecular dynamics calculations and molecular dynamics (MD) simulations using Matlantis were performed on the same input structure. Prior to the molecular dynamics calculations, structural relaxation was performed to determine the lattice volume. Furthermore, MD calculations were performed at a temperature of 1273 K under NVT ensemble (constant volume) conditions. The first-principles calculations were performed using 32 parallel cores. The detailed conditions for the first-principles calculations conform to the paper [1].

Results and Discussion
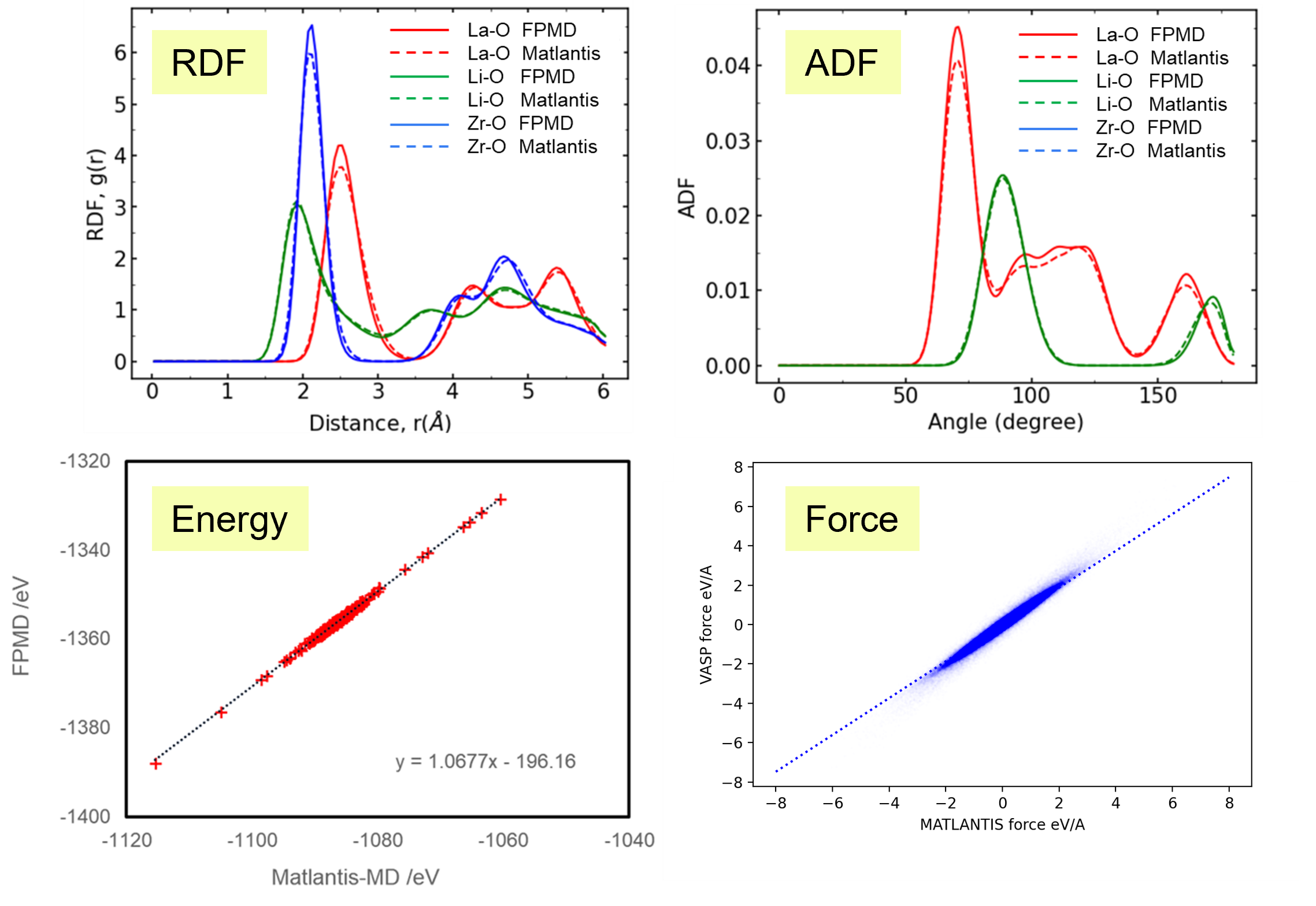
It takes tens of times longer to run the same number of MD steps using FPMD than using Matlantis, which demonstrated the high computation speed of Matlantis. Based on the ion trajectories obtained from both molecular dynamics calculations, the radial distribution function (RDF), which represents the distribution of distances between atoms, and the angular distribution function (ADF), which represents the distribution of bond angles defined by three ions, were compared. As a results, very good agreement was confirmed. In addition, the snapshots are extracted from the FPMD trajectories, and then the energy and forces acting on each atom was recalculated with Matlantis, so that we can compare the results with first-principles calculations. The diagnostic plots obtained showed that the two showed very good agreement. These results show that using Matlantis makes it possible to perform FPMD calculations at high speed. In this case, a small simulation cell consisting of 192 atoms was used to compare FPMD and Matlantis, so the difference in calculation speed was not large. However, it is expected that using Matlantis will make it possible to perform calculations within a realistic time even for large simulation cells that are impossible to achieve with FPMD.
In addition, it will be possible to evaluate the ionic conductivity of a large amount of garnet-type Li7La3Zr2O12 materials with simulations in a short time, which is expected to shorten the time required for composition optimization.
References
[1] R. Jalem, Y. Yamamoto, H. Shiiba, M. Nakayama, H. Munakata, T. Kasuga, K. Kanamura, “A Concerted Migration Mechanism in the Li Ion Dynamics of Garnet-Type Li7La3Zr2O12”, Chem. Mater., 25, 425-430 (2013) https://pubs.acs.org/doi/10.1021/cm303542xProfile of the case study provider
Nagoya Institute of Technology Nakayama Laboratory

tag
Publication date of this case: 2022.03.22
-




 URL
URL
Copied
what's new
NEW
Calculation and Verification of Methanol Synthesis Catalysts
Methanol synthesis catalystsLarge-scale screening
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis

SiO₂ Dry Etching Simulation Using LightPFP
Semiconductor
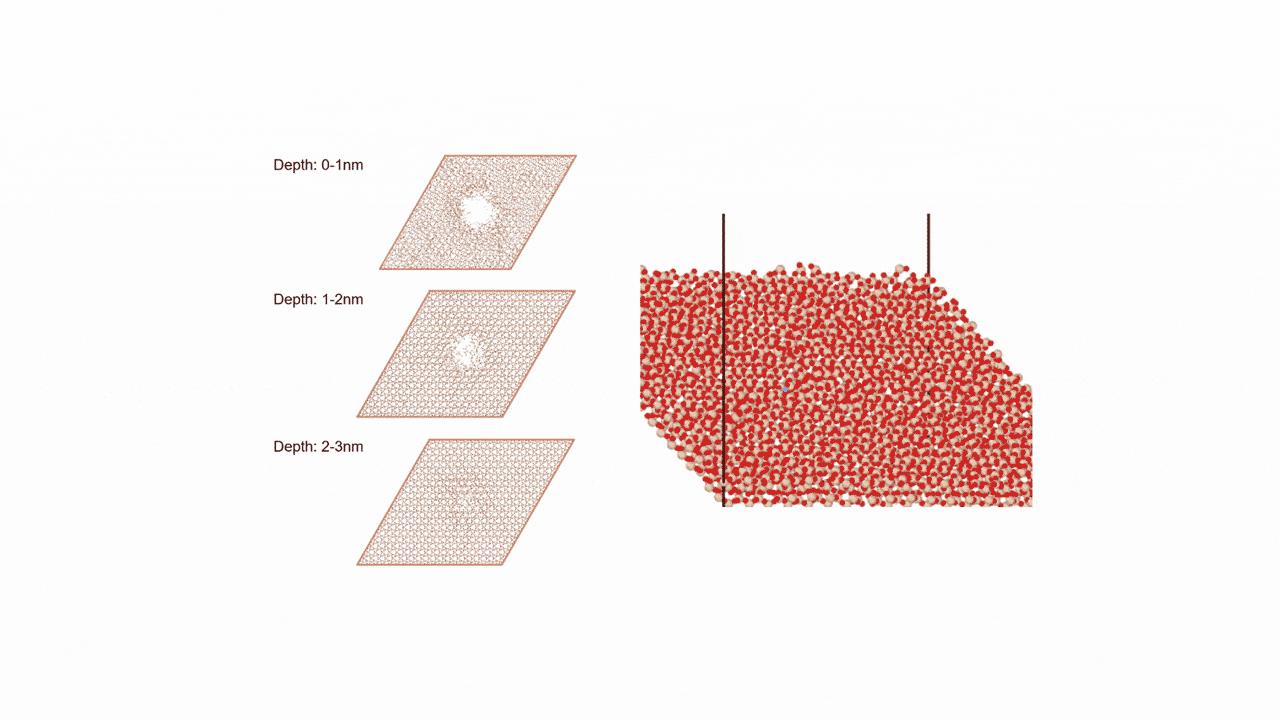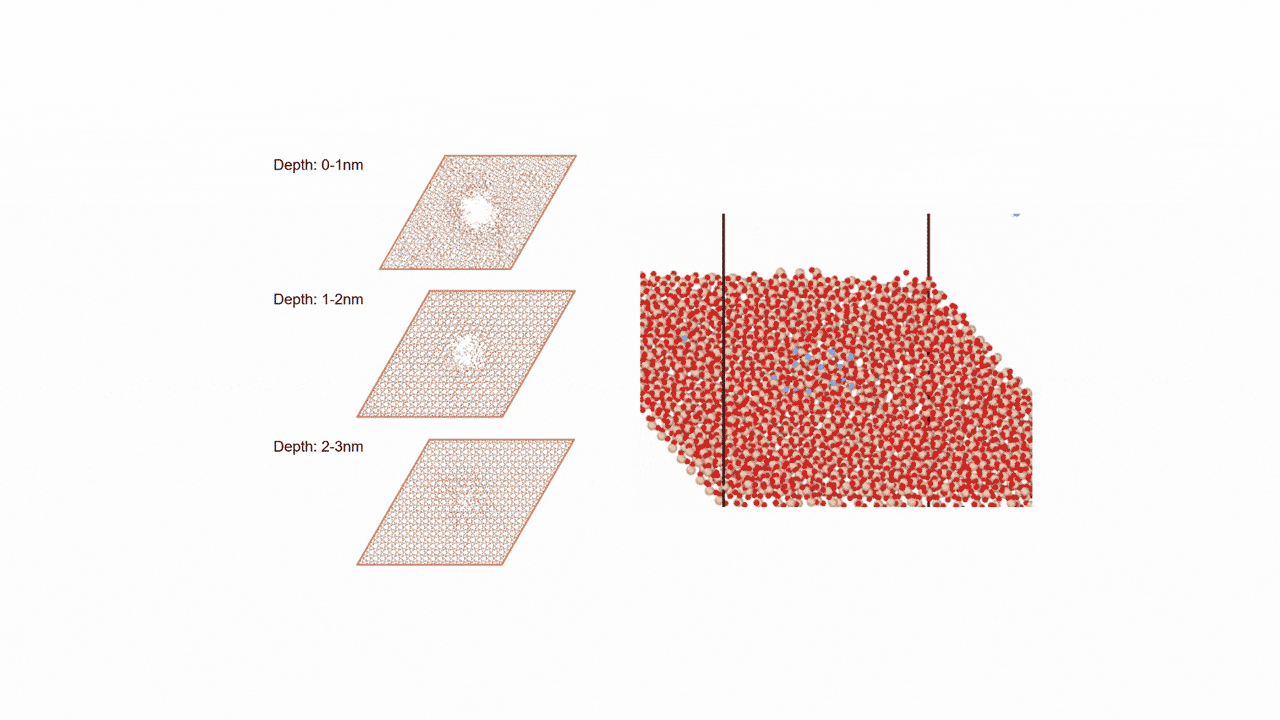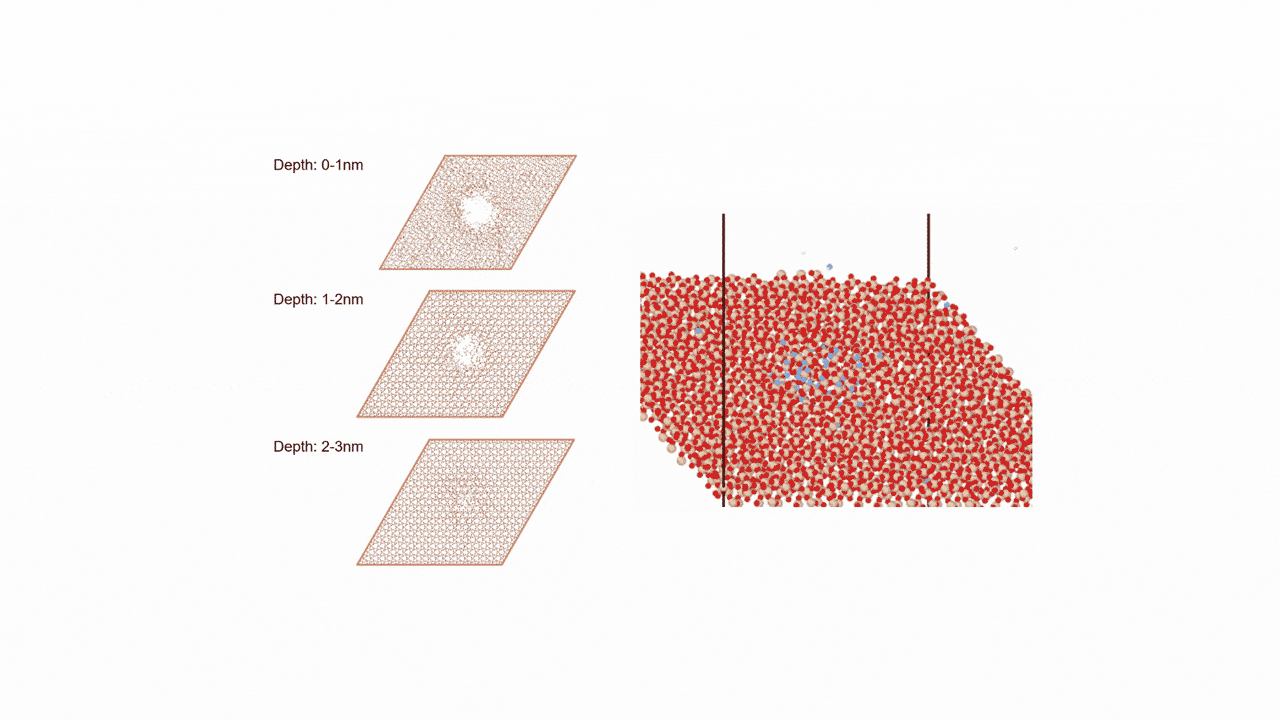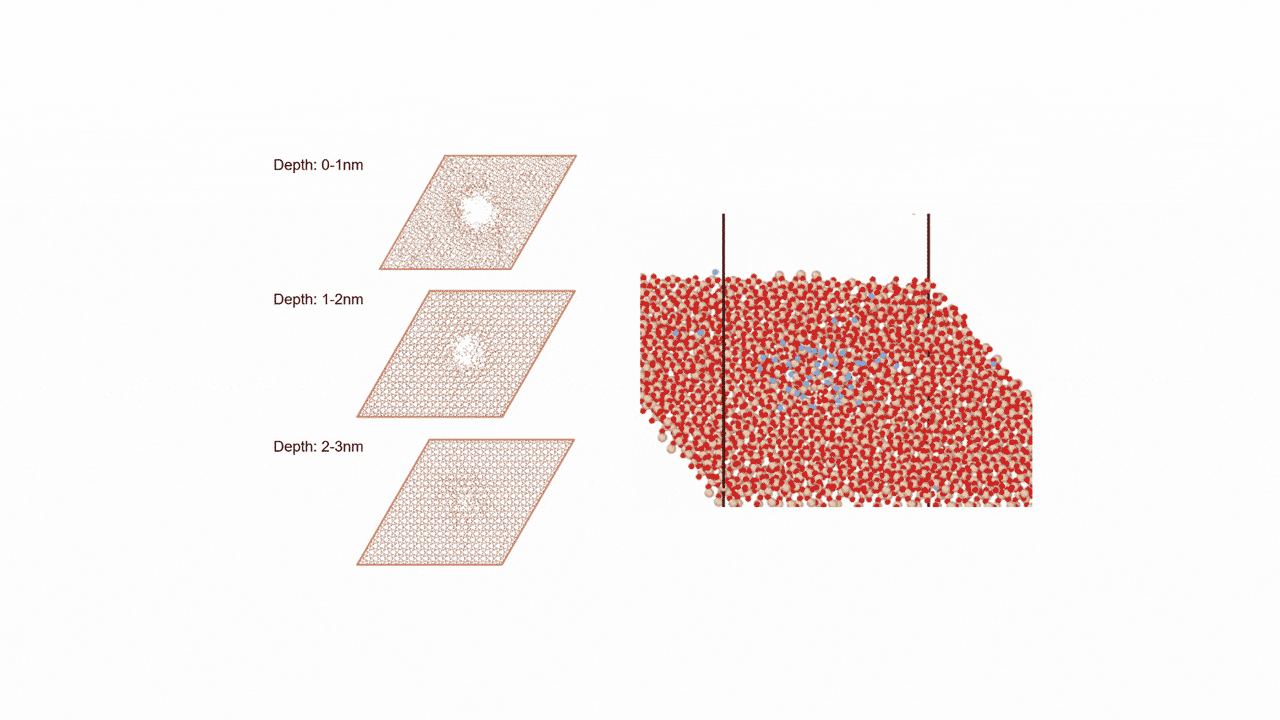
Analysis of Surface Reaction Mechanism between ALD Precursor and Substrate
SemiconductorsReaction Pathway Exploration