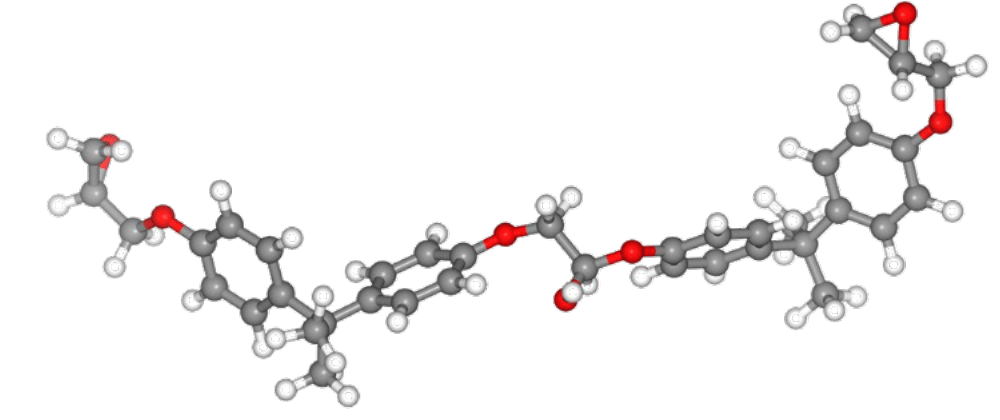
Thermal decomposition of epoxy molecules.
Introduction
The pyrolysis reaction of epoxy resins is important in the process design of plastics recycling. Typically, the pyrolysis of polymer molecules proceeds at high temperatures, resulting in the production of a variety of small molecules.
Such reaction processes can be simulated using classical molecular dynamics (MD) with a reactive force field (ReaxFF), which allows detailed analysis of decomposition reactions.
However, fitting the force field parameters of ReaxFF is difficult. To address this, we have performed calculations using PFP, a pre-trained universal potential, and compared the results with ReaxFF.
Calculation Models and Methods
A structural model consisting of 15 molecules of epoxy resin monomers was used for the simulations (see Figure 1). These molecules were equilibrated with the NPT ensemble. The system density of 0.95 g/cm³ obtained using PFP is in agreement with experimental results and previous studies using ReaxFF.
We then performed NVT simulations in which the temperature was gradually increased to evaluate the thermal decomposition reactions.
Results and Discussion
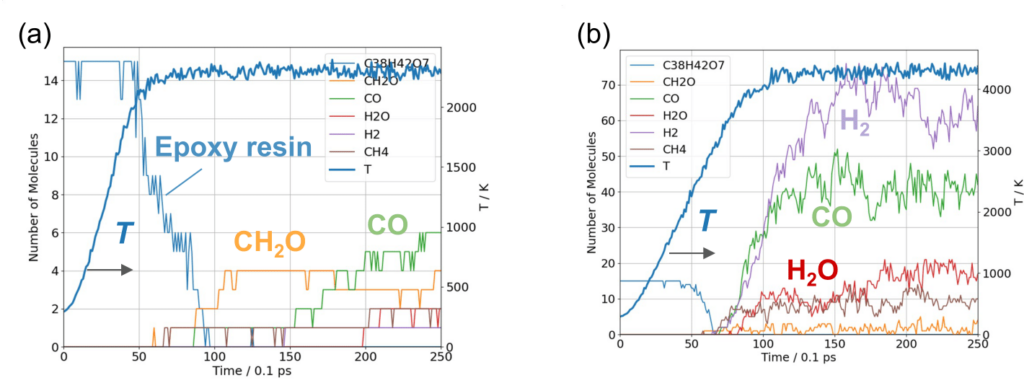
The number of molecules in the cell is plotted as a function of time in Figure 3. The results at a final temperature Tendof 2300 K (Figure 3(a)) show that the number of epoxy molecules decreases from about 2000 K, followed by the formation of small molecules. This suggests that there is an initial dissociation of ether bonds followed by thermal decomposition. The formation of small molecules occurs in the sequence CH2O, CO, then CH4
At a higher final temperature of Tend=4300 K, we observe the production of H2, CO, H2Oabove 2000 K (Figure 3(b)). Compared to the results at Tend=2300 K, the decomposition continues to smaller molecules.
These results are consistent with previous research and demonstrate that PFP can accurately calculate thermal decomposition of organic molecules.
Simulation Conditions

References
[1] Diao, Z.; Zhao, Y.; Chen, B.; Duan, C.; Song, S. Journal of Analytical and Applied Pyrolysis 2013, 104, 618-624tag
Publication date of this case: 2024.04.24
-




 URL
URL
Copied
what's new
Direct derivation of atomic displacement parameters using NNP-MD
Polymers

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
Polymers

SiO₂ Dry Etching Simulation Using LightPFP
Polymers
Analysis of Surface Reaction Mechanism between ALD Precursor and Substrate
Polymers
Catalyst Screening for Ammonia Synthesis Catalyst
Polymers