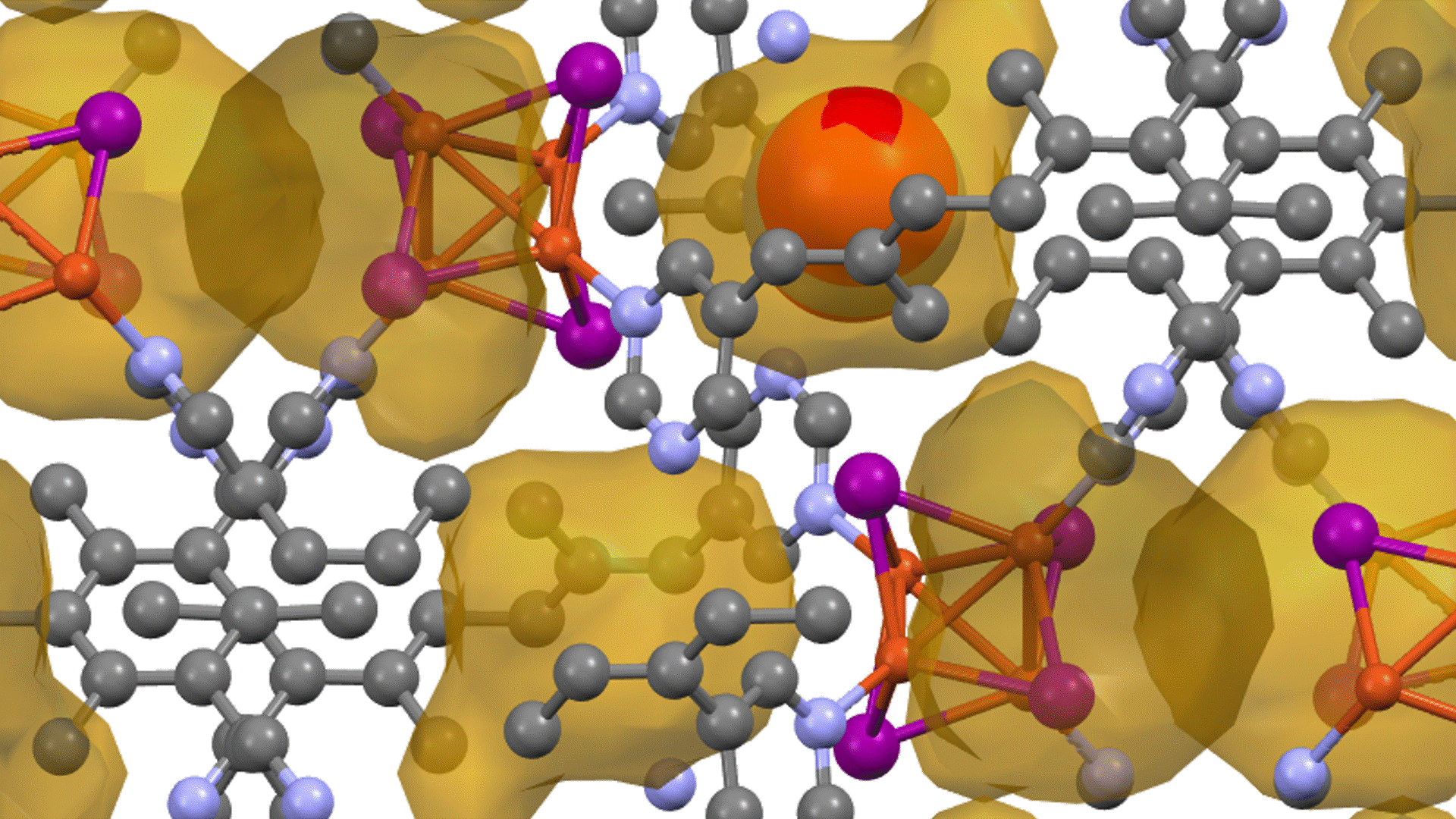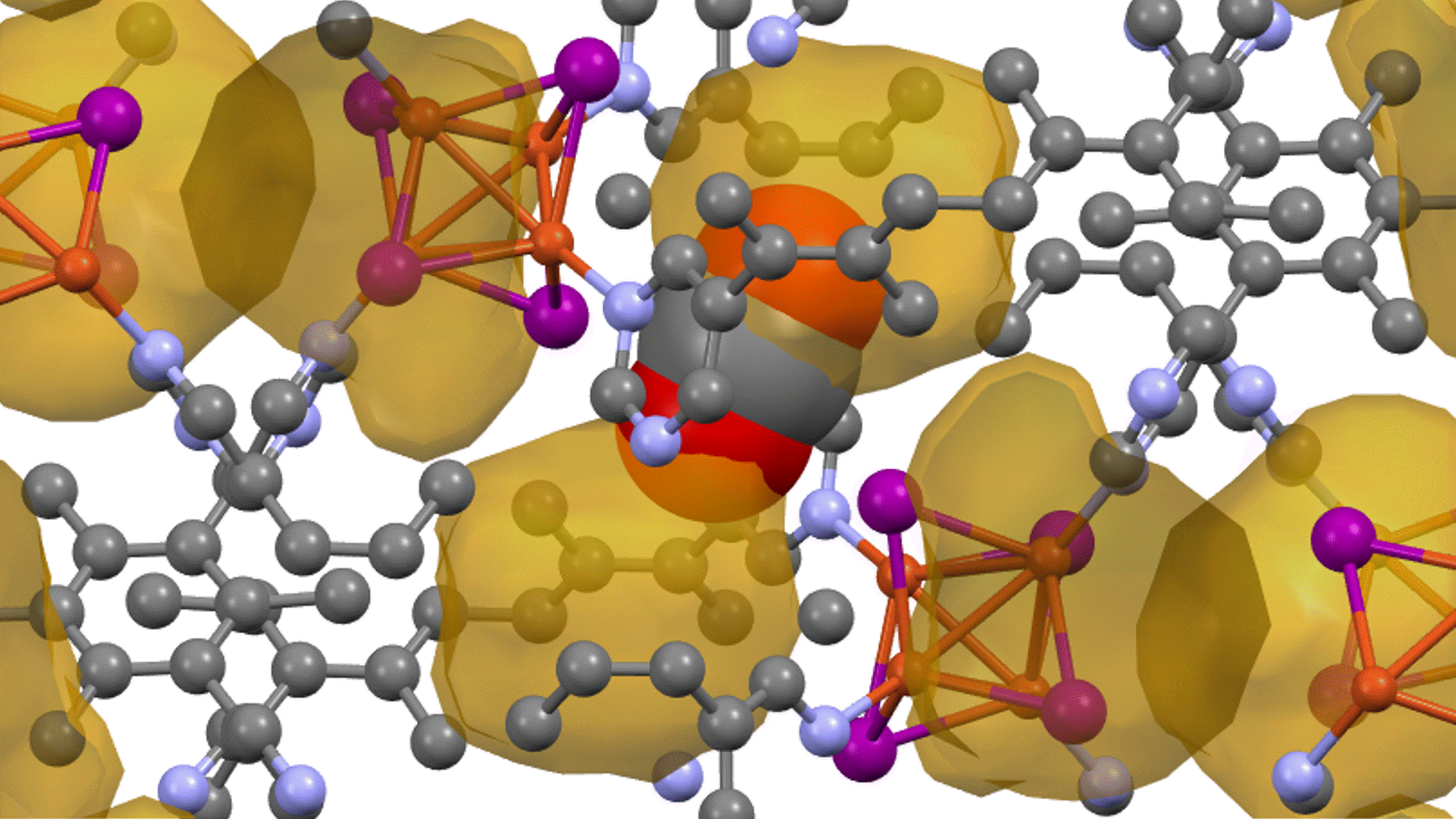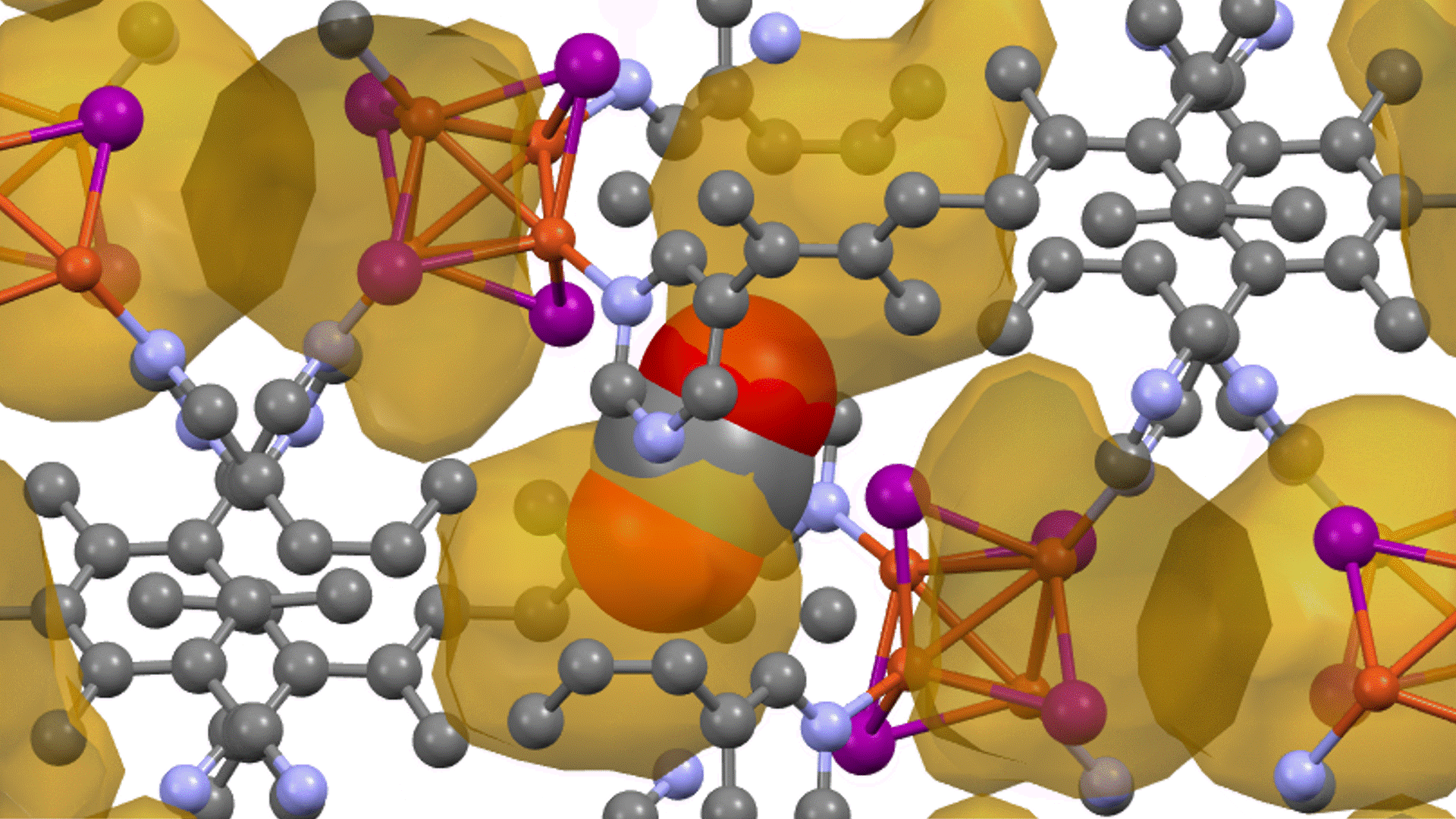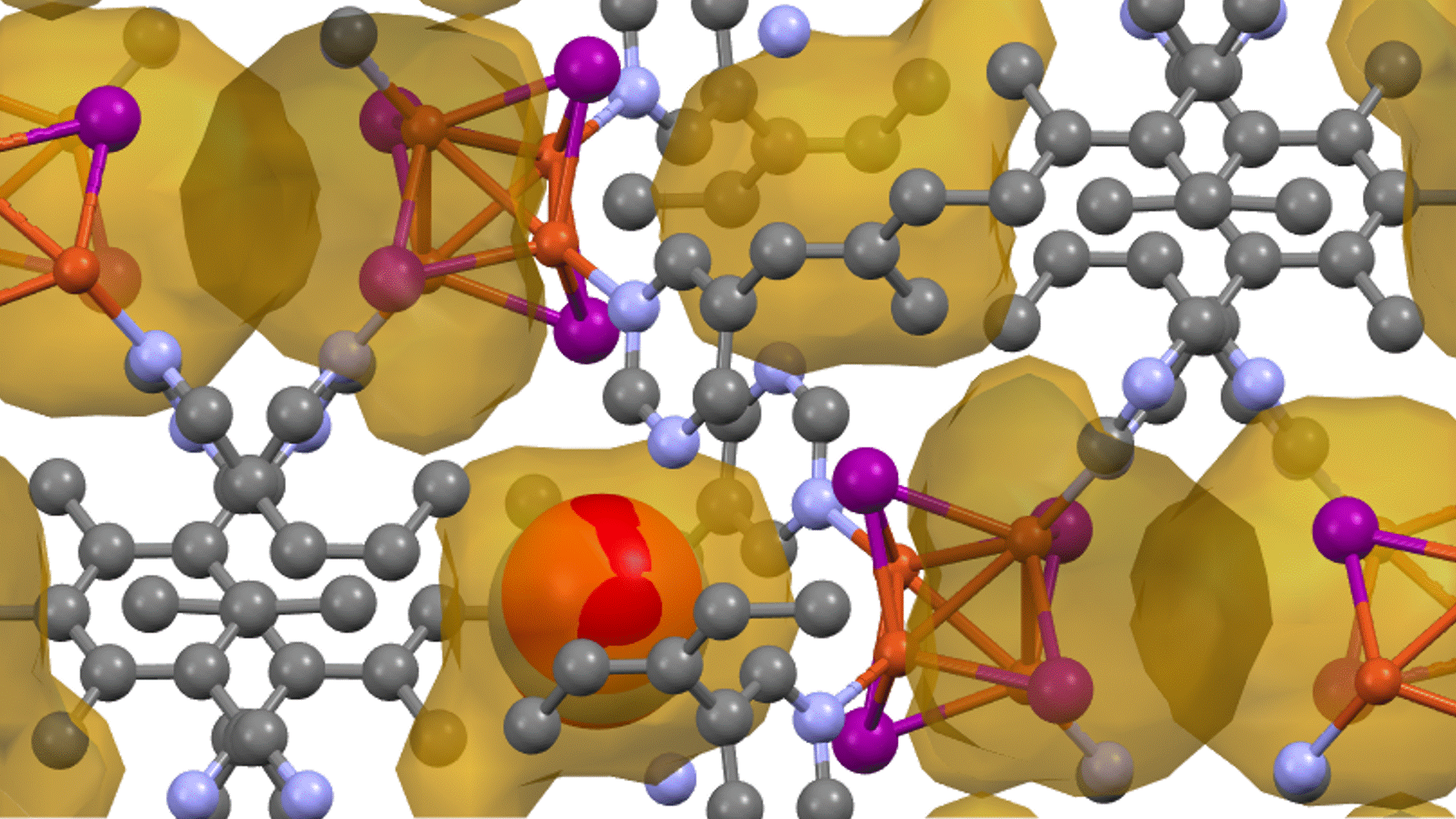
Direct derivation of atomic displacement parameters using NNP-MD
Case study provided by: National Institute of Advanced Industrial Science and Technology
Introduction
Atomic Displacement Parameters (ADPs), which describe the thermal vibration and positional fluctuation of atoms, play a crucial role in crystallographic structural analysis. While ADPs have conventionally been derived through lattice dynamics calculations, this approach faces difficulty when applied to complex crystals containing substitutional disorder or split sites. Therefore, this study[1] proposes a new approach for the direct derivation of anisotropic ADPs by combining Molecular Dynamics (MD) simulations with a machine-learned potential. The aim is to achieve a more accurate understanding of the dynamic atomic behavior in complex systems, such as thermoelectric materials, thereby providing valuable insights for structural analysis and material design. Furthermore, comparing the calculated ADPs with those from experimental crystal structure data (e.g., X-ray diffraction) allows for the validation of the analytical results. For MgO, used for method validation, applying a quantum mechanical correction for zero-point motion resulted in an agreement with experimental values within a few percent. For the thermoelectric material Ag8SnSe6, the deviation from experimental ADPs was generally less than 20%.
Calculation Models and Methods
In this study[1], Atomic Displacement Parameters (ADPs) by atom were directly derived from classical molecular dynamics (MD) simulations.The simulations were performed using Matlantis, which incorporates the universal machine-learned neural network potential (NNP), specifically the PreFerred Potential (PFP). This enabled the rapid calculation of highly accurate energies and forces, which are typically obtained from first-principles calculations (density functional theory). The calculations were conducted on MgO and various thermoelectric materials. The canonical, or constant number of atoms, volume and temperature (NVT), ensemble was employed with a Nosé–Hoover thermostat. MD simulations were performed for hundreds of thousands of steps.The anisotropic ADP was calculated as a tensor with components Uij , which represent the mean square value of the displacement from the atom's mean atom position. Specifically, these Uij components were obtained by computing the (co)variance matrix from the time average of the recorded atom positions. Furthermore, the temperature dependence of the ADP and the necessity of a zero-point motion correction were examined, confirming the effectiveness of the proposed method through comparison with experimental values.
Results and Discussion
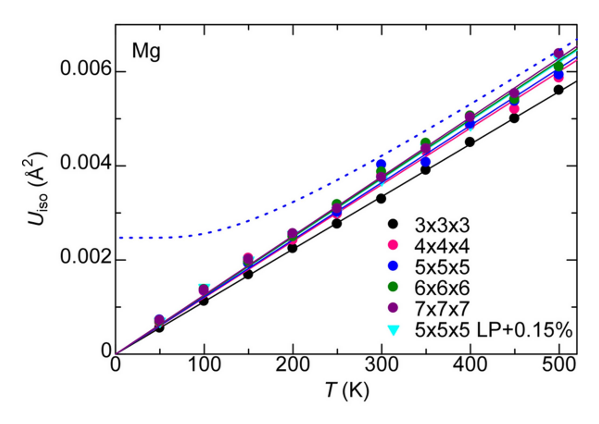
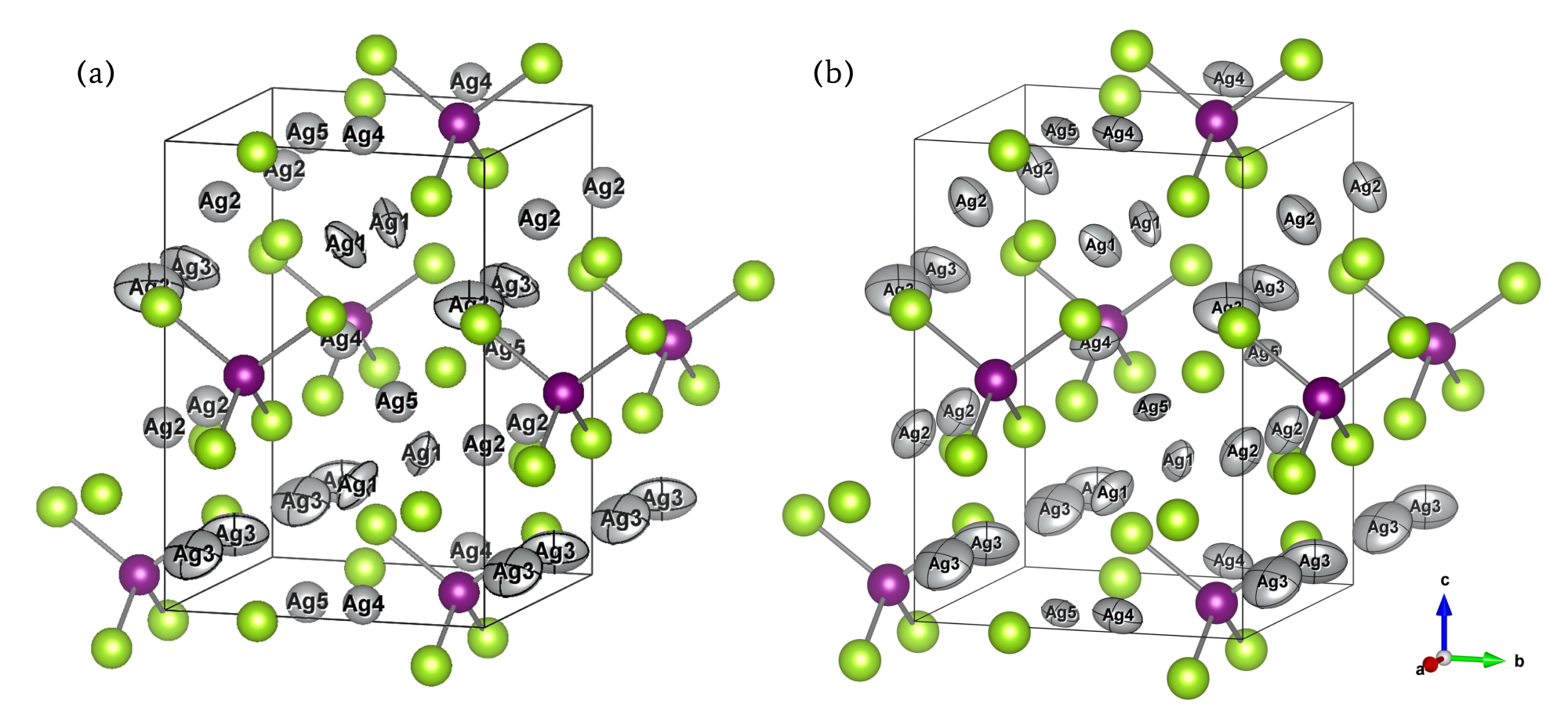
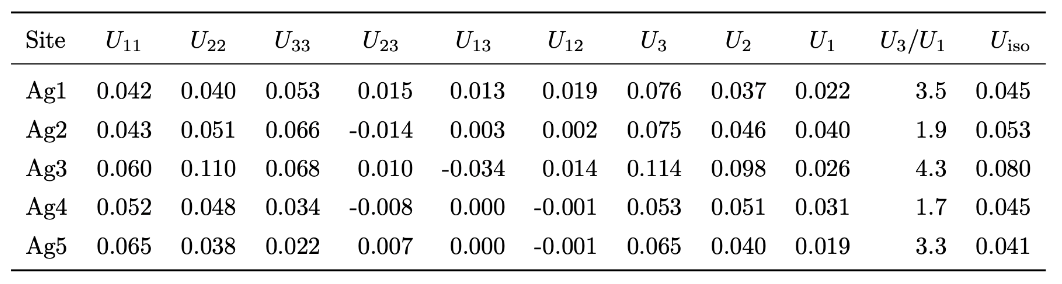
For MgO, the calculated ADP exhibited an almost linear dependence on temperature. When corrected for quantum mechanical effects (dashed line), the room-temperature result of 0.0042 Ų showed good agreement with the experimental value from X-ray diffraction (XRD) of 0.0038 – 0.0040 Ų [2] (Fig. 1). In Ag₈SnSe₆, the large anisotropy and "rattling" behavior of the Ag atoms were successfully reproduced, suggesting the possibility of site splitting (Fig. 2). At 300 K, U3/U1(the ratios of the major to minor semi-axes of the displacement ellipsoid) for the highly anisotropic Ag3 and Ag1 sites showed relative errors of -15.8% and -17.6%, respectively, against the experiment. The isotropic ADP Uiso, representing the overall magnitude of the displacement ellipsoid, showed relative errors of -5% and -12% (Table 1).The paper [1] also presented results for other thermoelectric materials.
In Na2In2Sn4, significant atom migration was observed above 200 K, leading the calculated ADP to significantly overestimate the experimental value, thus clarifying the necessity to account for the diffusion behavior. For BaCu1.14In0.86P2, a large discrepancy was confirmed between the ADP "by atom" and "by site" due to the disorder at the Cu/In site.
Future challenges include improving statistical precision through longer simulations and larger supercells, and the direct incorporation of zero-point motion to account for quantum effects. Applications are anticipated in utilizing the anomalous behavior of ADPs to enhance thermoelectric performance and to refine crystal structures. This method opens up new possibilities for the structural analysis of materials, including those with substitutional disorder and split sites, which were previously difficult to study, establishing it as a powerful tool for future material design and functional exploration.
Table 1: U of Ag in Ag₈SnSe₆ at 300 K [Å2]
Simulation Conditions
| item | Details |
|---|---|
| Potential | PFP v7.0.0 CRYSTAL |
| Simulation Conditions | MgO 50,000 , 2 fs time step, 1000 atoms Ag8SnSe6:60,000 steps, 2 fs time step, 810 atoms |
| Temperature | MgO: 50-500K Ag8SnSe6: 50-300K |
| Ensemble | NVT |
References
[1] Hinuma, Y., Acta Cryst, A81, 279 (2025) DOI 10.1107/S2053273325004620 [orcid.org], [journals.iucr.org]
[2] Lawrence, J. L., Acta Cryst. A29, 94 (1973); Sasaki, S., et al., Proc. Jpn. Acad. Ser. B 55, 43 (1979); Tsirelson V. G., et al., Acta Cryst. B54, 8 (1998)
[3] Takahashi, S., et al., Cryst. Growth Des. 24, 6267 (2024)
Profile of the case study provider
National Institute of Advanced Industrial Science and Technology

Publication date of this case: December 16, 2025
-




 URL
URL
Copied
what's new
A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
Atomic Displacement ParametersThermoelectric Materials

SiO₂ Dry Etching Simulation Using LightPFP
Atomic Displacement ParametersThermoelectric Materials
Analysis of Surface Reaction Mechanism between ALD Precursor and Substrate
Atomic Displacement ParametersThermoelectric Materials
Catalyst Screening for Ammonia Synthesis Catalyst
Atomic Displacement ParametersThermoelectric Materials
Analysis of CO2 adsorption dynamics of MOF using NEB method
Atomic Displacement ParametersThermoelectric Materials