PFP Descriptors
~~~Taking on previously incalculable characteristics with machine learning~~~
What are PFP Descriptors?
PFP, a universal machine learning potential (uMLIP), can calculate the properties of various materials. However, even with PFP, there are properties that are difficult to calculate due to the time and spatial scales involved. PFP Descriptors are a new option for predicting these properties.
In the graph neural network used in PFP, each atom is assigned a vector that represents the local environment around it. These vectors are extracted and called PFP Descriptors. They can be referenced as a 256-dimensional array per atom. PFP Descriptors are trained representations for predicting energy and force, but they potentially incorporate information about interactions between atoms, making them suitable for use as descriptors in machine learning.
For example, MLIP achieves high-speed energy calculations by omitting the process of electronic state calculations in quantum chemical calculations. Therefore, it has been difficult in principle to directly calculate physical properties that depend on electronic states in simulations using MLIP. In contrast, PFP Descriptors are thought to acquire information about the chemical environment of atoms in the process of learning about energy and forces. By using this in conjunction with machine learning, it is expected that highly accurate predictions of properties related to electronic states, which have been difficult to achieve with MLIP until now, will become possible.
*This function is a trial service, and the terms of use may be changed, incompatible changes may be made, or the service may be discontinued without notice.
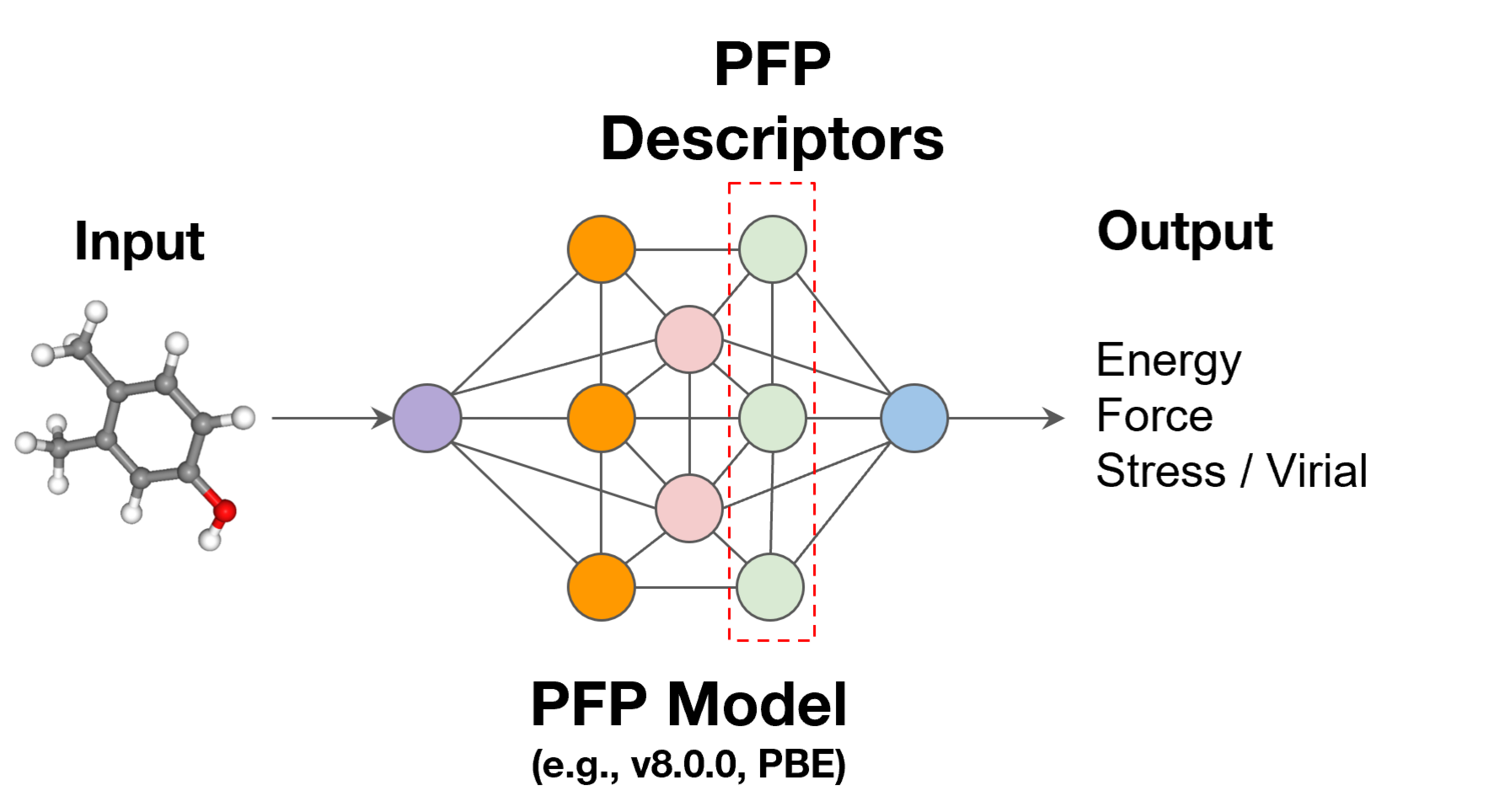
What you can do with PFP Descriptors and its benefits
- Generation of atomic features that reflect the local environment
- High-precision prediction of atomic and material properties
- Analysis of changes in the state of atoms and materials
Applications
Atomic-level utilization
NMR Chemical shift prediction
PFP Descriptors express the local environment of each atom as a vector. Because the chemical shift in NMR strongly depends on the local environment of that atom, we confirmed that by using PFP Descriptors, which can precisely express the local environment, in machine learning, the chemical shift in NMR can be predicted with high accuracy.
(Reference: GitHub)
Use on a material-by-material basis
Physical property prediction
PFP Descriptors are also effective for predicting the properties of entire materials. They have achieved high prediction accuracy for a wide range of properties included in the public benchmark (Matbench). PFP is pre-trained on a vast amount of material data, and a wealth of information about materials is stored in its intermediate layer. By using PFP Descriptors extracted from this intermediate layer to build a machine learning model, we believe we were able to create a highly accurate prediction model.
(Reference: The Power of PFP Descriptors)
Trajectory dimensionality reduction
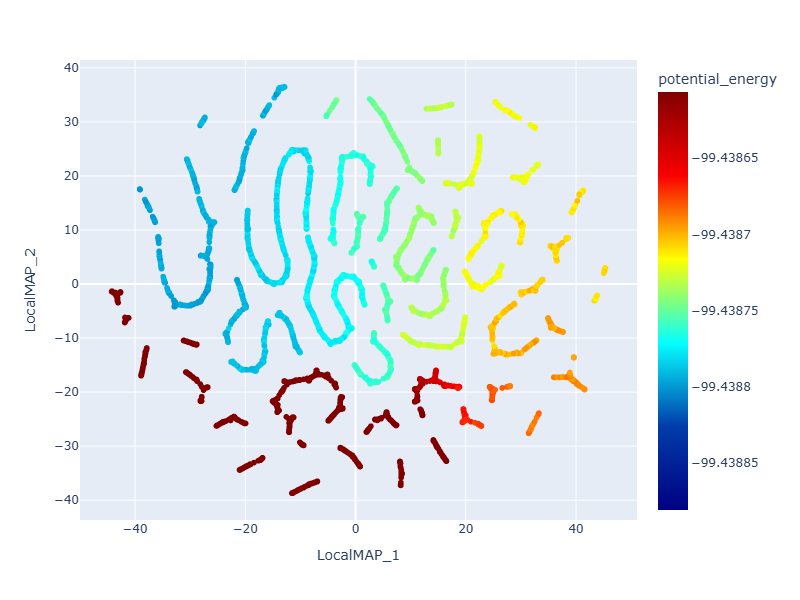
During the process of structural optimization or molecular dynamics simulation, materials undergo various structural changes. Understanding these structural changes is important, but conventional methods require manual definition of physical quantities such as bond lengths and bond angles for visualization. However, by using PFP Descriptors, it is possible to vectorize the structure at each point in the simulation and reduce the dimension, making it easy to understand the similarities between structures.
The figure below shows the dimension reduction results for the trajectory in the geometry optimization calculation of L-alanyl-L-alanine using PFP Descriptors. As can be seen from the figure, structures with similar potential energies are placed close to each other on the plot, and the structure can be visualized appropriately based on its similarity.
In addition, PFP Descriptors can be used for various purposes, such as interpreting machine learning models. If you are interested, please feel free to contact us.
-




 URL
URL
Copied