Past News
Conferences and lectures
Walter E. Washington Convention Center2025.8.17-21 (USA)
Event Announcement: Matlantis at ACS Fall 2025
Matlantis Corporation will be exhibiting at ACS Fall 2025, which will be held from August 17th to August 21st, 2025 (EDT). Two members of Matlantis will be giving poster presentations and will also have a booth. In addition, there will be one poster presentation related to Matlantis from an external organization. Please see below for details.
Click here for this event report
Exhibition and booth information
Exhibition period: August 17th - August 21st, 2025 (EDT)
Location: Walter E. Washington Convention Center, 801 Allen Y. Lew Place NW, Washington
Booth: 2045
Presentation Information
Room: Hall C (Walter E. Washington Convention Center)
Speaker 1: Hirotaka Yonezawa
Session: Poster Board #704
Date and Time: August 20, 2025, 6:00 PM – 8:00 PM (EDT)
Division: Division of Chemical Information (CINF)
Session Type: Poster Presentation
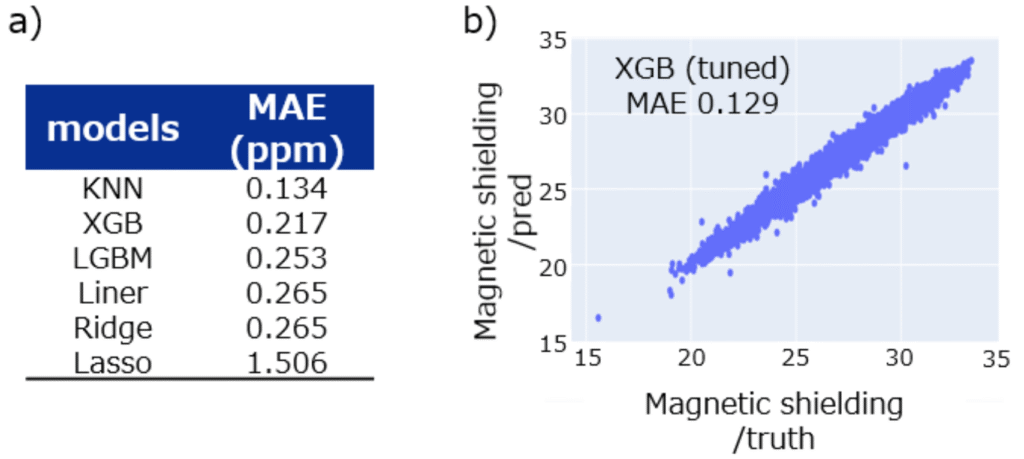
Title: NMR chemical shift prediction with PFP descriptor: An application of universal machine learning interatomic potential
Overview: Nuclear Magnetic Resonance (NMR) is a crucial technique for identifying the structure of organic molecules. Especially for complex molecules, it is essential to compare simulated spectra with measured data to assign the peaks and determine the structure accurately. This is why the research of chemical shift prediction has long been conducted.
Approaches to chemical shift prediction divided can be categorized into two types: ab initio techniques and data-driven methods. The latter can be further into three categories: additive increment-based, HOSE code, and machine learning.
Approaches have shown comparable good performance. However, these techniques share a common limitation: the representation of the surrounding environment beyond directly connected atoms.
In this study, we propose the approach to create NMR chemical shift prediction models with the descriptors extracted from machine learning interatomic potentials (MLIPs). This method is superior to previous methods in terms of representing the environment around atoms, especially geometrically distant atoms. This is because MLIPs already account for various intra/inter- and short/long-range atomic interactions and the descriptors can represent the complex interactions and local environments of atoms, providing a comprehensive basis for shift chemical prediction.
Hirotaka Yonezawa
Customer Success Engineer, Matlantis Corporation.
After obtaining his PhD in Science from the University of Tokyo in 2018, he worked at a consulting firm, improving business efficiency through data science. He joined Matlantis in 2024. He aims to create a world where even experimental chemists can improve the efficiency of their research through computation. His specialties are organic synthesis, supramolecules, and photochemistry.

Speaker 2: Akihiro Nagoya
Session: Poster Board #928
Date and Time: August 19, 2025, 6:00 PM – 8:00 PM (EDT)
Division: Division of Computers in Chemistry (COMP)
Session Type: Poster Presentation
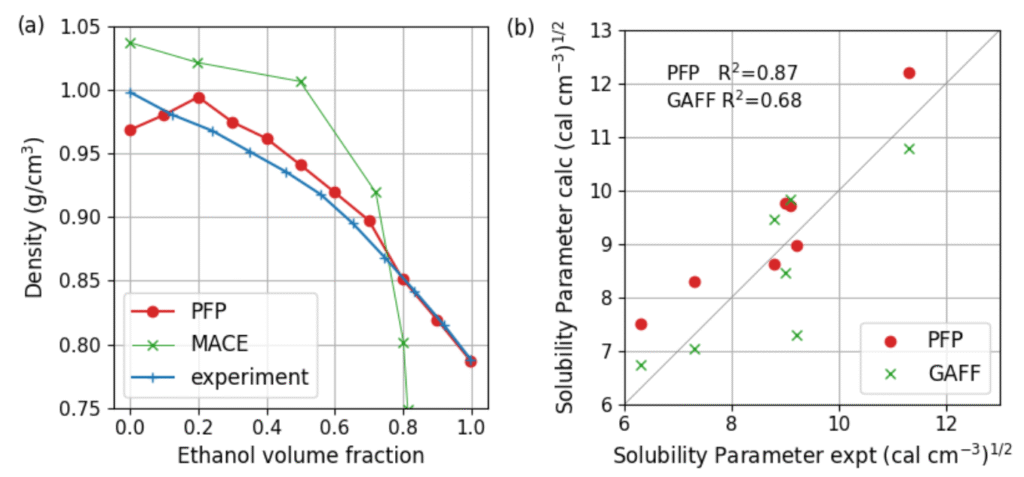
Title: Application of a universal graph neural network potential for molecular dynamics of liquid phases
Overview: Molecular dynamics (MD) simulations are essential tools for studying the structure and dynamics of liquid phase in chemistry, materials science and biophysics. Applications include solvation structure analysis, ion transport in electrolytes, phase equilibria, interfacial behavior and diffusion processes. However, classical force fields often lack the accuracy required to capture complex interactions, such as chemical reactions at solid/liquid interface, while ab initio MD is too computationally expensive.
Recent advances in machine learning potentials (MLPs) have opened new possibilities for liquid phase simulations. MLPs have been successfully applied to a number of liquid systems achieving DFT accuracy at much lower cost. MLPs allow MD simulations of complex phases that are inaccessible using quantum methods. On the other hand, the atomic configuration may be outside the training data set during long MD simulations, leading to error accumulation or inaccurate physical behavior. The Preferred Potential (PFP), integrated into Matlantis™, is a recently developed universal graph neural network (GNN) potential. PFP achieved a high level of accuracy and robustness due to its training on the huge DFT dataset. It offers superior applicability, transferability, and consistency across a broad range of systems without fine-tuning for specific systems. Thus, it is widely used for various applications such as catalysts, battery materials, metals and polymers.
In this study, we validated the accuracy of PFP for liquid phases. The solubility of organic liquids was evaluated using the Hildebrand solubility parameter derived from MD simulations. PFP agrees well with experimental data and predicts solubility more accurately than the GAFF force field. Furthermore, we validated the density of a water-ethanol mixture, with PFP showing good agreement with experimental data. Note that the underestimation for pure water is due to known errors in the PBE data set. It can be improved by training the model with more sophisticated functional. The results demonstrate that PFP is suitable for practical MD simulations and facilitates materials development.
Akihiro Nagoya
Senior Applications Scientist, Matlantis Corporation.
After graduating from Osaka University, he worked as a researcher at Toyota Central R&D Labs., Inc. For approximately 15 years, he was involved in research on first-principles calculations for solar cell materials, fuel cell platinum catalysts, and two-dimensional materials, as well as MI using classical MD calculations for polymers. He then worked at the Central Technical Research Laboratory of ENEOS Corporation before joining Matlantis. His current interest is in applications of machine-learning potential to battery materials and metal materials.

External Collaborators’ Presentation
2025年8月18日
Session: Chemical Reaction Networks, Retrosynthesis, and Reaction Prediction
Date and Time: August 18, 2025, 5:20 PM – 5:40 PM (EDT)
Division: Division of Computers in Chemistry (COMP)
Room: Hall E – Room 25 (Walter E. Washington Convention Center)
Session Type: Oral - In-person
Title: Automated generation of reaction pathways using neural networks
Presenters: Akihide Hayashi, So Takamoto, Ju Li, Hirotaka Akihide, Daisuka Okanohara
Organization: Preferred Networks, inc. & Massachusetts Institute of Technology
2025年8月19日
Session: Poster Board #922
Date and Time: August 19, 2025, 6:00 PM – 8:00 PM (EDT)
Division: Division of Computers in Chemistry (COMP)
Room: Hall C (Walter E. Washington Convention Center)
Session Type: Poster Presentation
Title: r2SCAN level universal neural network potential for molecules, crystals and surfaces
Presenters: Chikashi Shinagawa, So Takamoto, Daiki Shintani, Katsuhiko Nishimra, Kohei Shinohara, Shigeru Iwase, Yuta Tsuboi, Ju Li
Organization: Preferred Networks, inc. & Massachusetts Institute of Technology
For official information about the conference, please click here (external site) >> ACS Fall 2025
公開日:2025.08.13
-




 URL
URL
Copied
New Events and Seminars
NEW
Upcoming events
Webinars
Online2026.03.17(JST)
Identifying Unknown Crystal Structures Prior to Experiments — Designing and Applying Crystal Structure Prediction with Matlantis CSP —

NEW
Upcoming events
Conferences and lectures
Institute of Science Tokyo Ookayama Campus & Online2026.3.15-18
Presented at the 73rd Spring Meeting of the Japan Society of Applied Physics

NEW
Upcoming events
Webinars
Hybrid event held on-site (Akihabara UDX) and online (Zoom)on February 25, 2026
Presentation at the 21st Materials Workshop | The true value and future prospects revealed through the thorough use of Matlantis

Past News
Webinars
zoom2026/2/16
Matlantis Webinar|Unveiling Semiconductor Processes at the Atomic Level~The Cutting Edge of AI Simulation~

Video Streaming
Webinars
OnlineOn-Demand
[On-Demand Webinar] Matlantis: Universal Atomistic Simulation for AI-Driven Materials Discovery
