Resource Library
PFP v7.0.0の検証
概要
- PFP v7.0.0で新たに対応した24元素について、力およびエネルギーの再現性を確認しました。
- PFP v7.0.0では有機結晶と錯体結晶に関して、安定構造付近の結晶のエネルギー・力の再現性が向上しました。また体積および格子定数の再現性が向上しました。
- PFP v7.0.0とv6.0.0とでは、液体の密度の再現性は概ね同等でした。ただし、比較的長鎖のパーフルオロアルカンの密度を過小評価する傾向があり、原因を調査中です。
- PFP v7.0.0ではv6.0.0と比較して、結晶構造のエネルギーの順序の再現性がわずかに悪化しており、原因を調査中です。
- PFP v7.0.0は、無機結晶に関する高密度構造におけるポテンシャルエネルギー曲面の安定性が改善しました。一方で有機結晶および錯体結晶に関してはわずかに悪化していますが、問題にはならない程度と考えています。
COD構造によるベンチマーク
PFPの現実世界に存在する多様な物質に対する再現性を確認するため、Crystallography Open Database (COD) 1を利用して再現性の検証を行いました。なお、ここで用いるCOD構造を用いたベンチマークデータセットはPFPの学習には利用していません。
ベンチマークデータセット
CODから、PFP v7.0.0以降でサポートする96元素のみで構成される結晶構造のうち、単位胞の体積が2200ų以下のものを取得しました。これはv3.0.0から使用していた72元素対応のベンチマークデータセットを拡張したものです。ただし、各サイトの占有率が0.99未満のものや、対称性の指定に問題があるもの等は除外しています。これらの結晶構造に対して、構造最適化とサイト微小変位を加えた構造を用意し、DFT計算を行いベンチマークデータセットを生成しました。DFT計算の計算条件はCRYSTAL_U0に相当するものですが、構造最適化を安定させるためSCFの収束条件(EDIFF)を10-7とより厳しくしています。CRYSTAL_U0の計算条件に関しては、About PFPを参照してください。
PFP v5.0.0以降では、CODに含まれる結晶構造の一部を学習データセットとして用いています。このため、本検証にあたってはこれらの結晶構造を除外しています。ただし、これによって除外されなかった結晶構造についても、学習用データセットに同じ結晶構造が含まれている場合もあります。例えば、Materials Project 2 に同じ結晶構造が登録されており、この構造を学習用データセットでも利用している場合があります。
構造最適化
比較対象とするDFT条件での構造を得るために、格子定数(長さと角度)を含めた構造最適化をVASPで行いました。構造最適化の収束条件は、forceが0.03eV/Å以下としました。
データセットには、構造最適化後の構造のみが含まれます。
微小変位
安定構造周辺の局所的なエネルギー曲面の再現性を確認するため、構造最適化後に微小変位を加えました。詳しくは、PFP論文3のSupporting Information, NOTE 10にある“site position displacement”を参照ください。ただし、変位の大きさは論文中よりも小さくしています。
追加のサポート元素
PFP v7.0.0では、新たに24の元素がサポート元素に追加されました。これらの元素のうち少なくとも1つを含む構造に対する結果です。
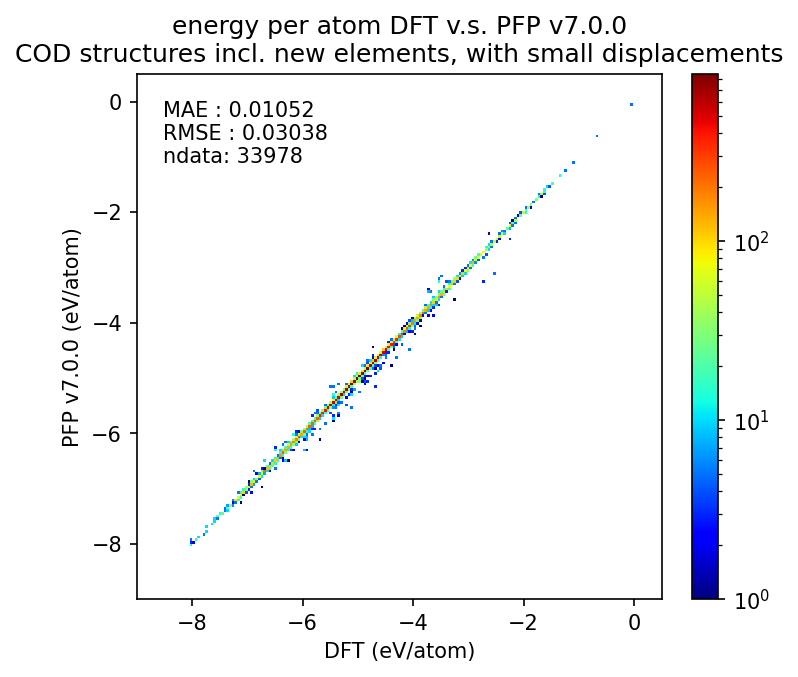
以下は微小変位を加えた構造に対する、エネルギーと力のDFT結果の再現性についての図です。エネルギーに関しては1原子あたりのエネルギーを比較し、力に関しては各原子にかかる力のxyz成分を独立の点としてプロットしています。
| エネルギー | 力 |
|---|---|
 |
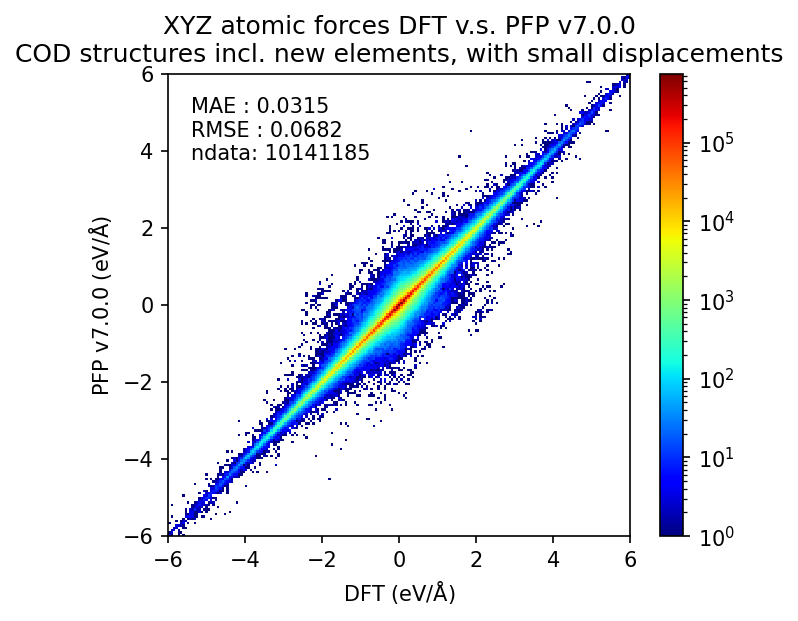 |
詳細は後述しますが、従来のサポート元素のみを含む無機結晶構造に対するエネルギーと力のMAEはそれぞれ0.058 eV/atom、0.24 eV/Åです。追加のサポート元素に対するMAEはこれより大きいものの、乖離してはいません。このため、PFP v7.0.0ではエネルギーと力ともに新元素に対応したと言えます。
PFP v7.0.0での新しい元素のサポートは、主に結晶構造を対象としており、表面構造などのデータは限られています。このため、今回追加された元素に対する、表面構造などの再現性は不十分である可能性があることに留意してください。
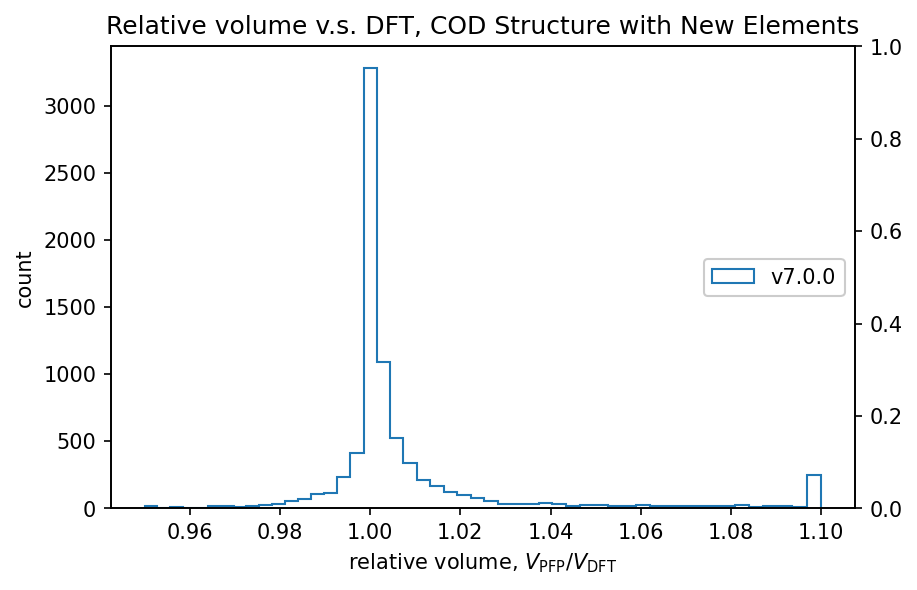
以下は体積についてのDFT結果の再現性についての図です。詳細に関しては「従来のサポート元素」内の「体積と格子定数の再現性」の項目を参照してください。
| 相対体積 | 相対誤差 |
|---|---|
 |
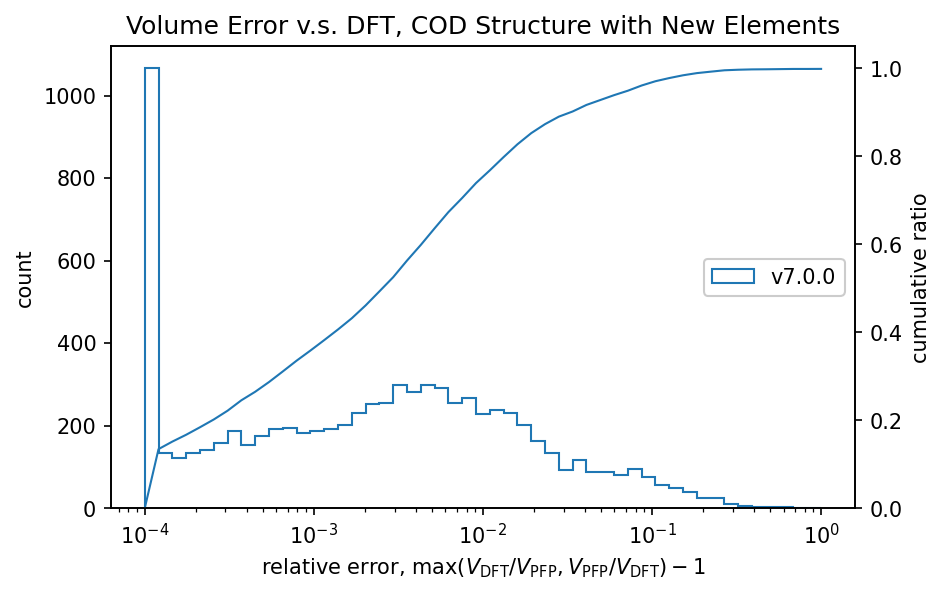 |
詳細は後述しますが、従来のサポート元素のみを含む無機結晶構造についてはそのうちの90%の系において、DFTとPFPでの体積の相対誤差が0.03以下に収まります。追加のサポート元素に対するこの相対誤差の値はこの値と同等であり、体積に関してもPFP v7.0.0は新元素に対応したと言えます。
従来のサポート元素
PFP v6.0.0でサポートされていた72元素のみを含む構造に対する結果です。
エネルギーと力の再現性
微小変位を加えた構造を対象として、エネルギーと力の再現性をPFP v7.0.0とPFP v6.0.0で比較しました。比較しやすくするため、プロット範囲を固定しており、範囲外にも比較的少数のデータ点が存在することに注意してください。
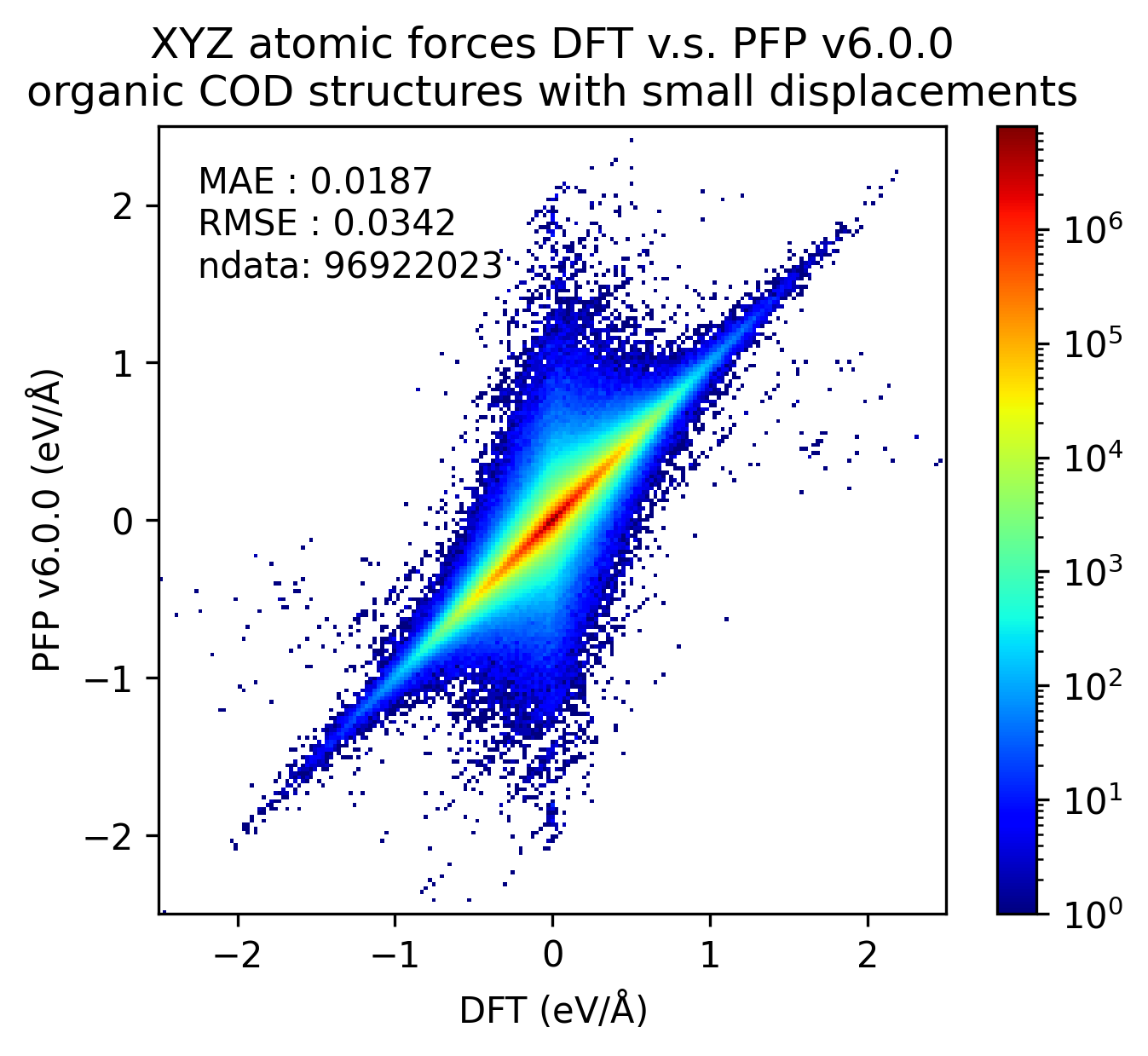
有機結晶
H, C, N, O, P, S, F, Cl, Br, Iのみで構成される構造を有機結晶とみなして、比較をしました。
y-yプロットではエネルギーは1原子あたりで比較し、力は各原子にかかる力のxyz成分を独立に比較しています。
ヒストグラムでは1原子あたりのエネルギーの誤差の絶対値、および、各原子にかかる力の誤差のL2ノルムを示しています。
| v7.0.0 | v6.0.0 | histogram | |
|---|---|---|---|
| エネルギー | 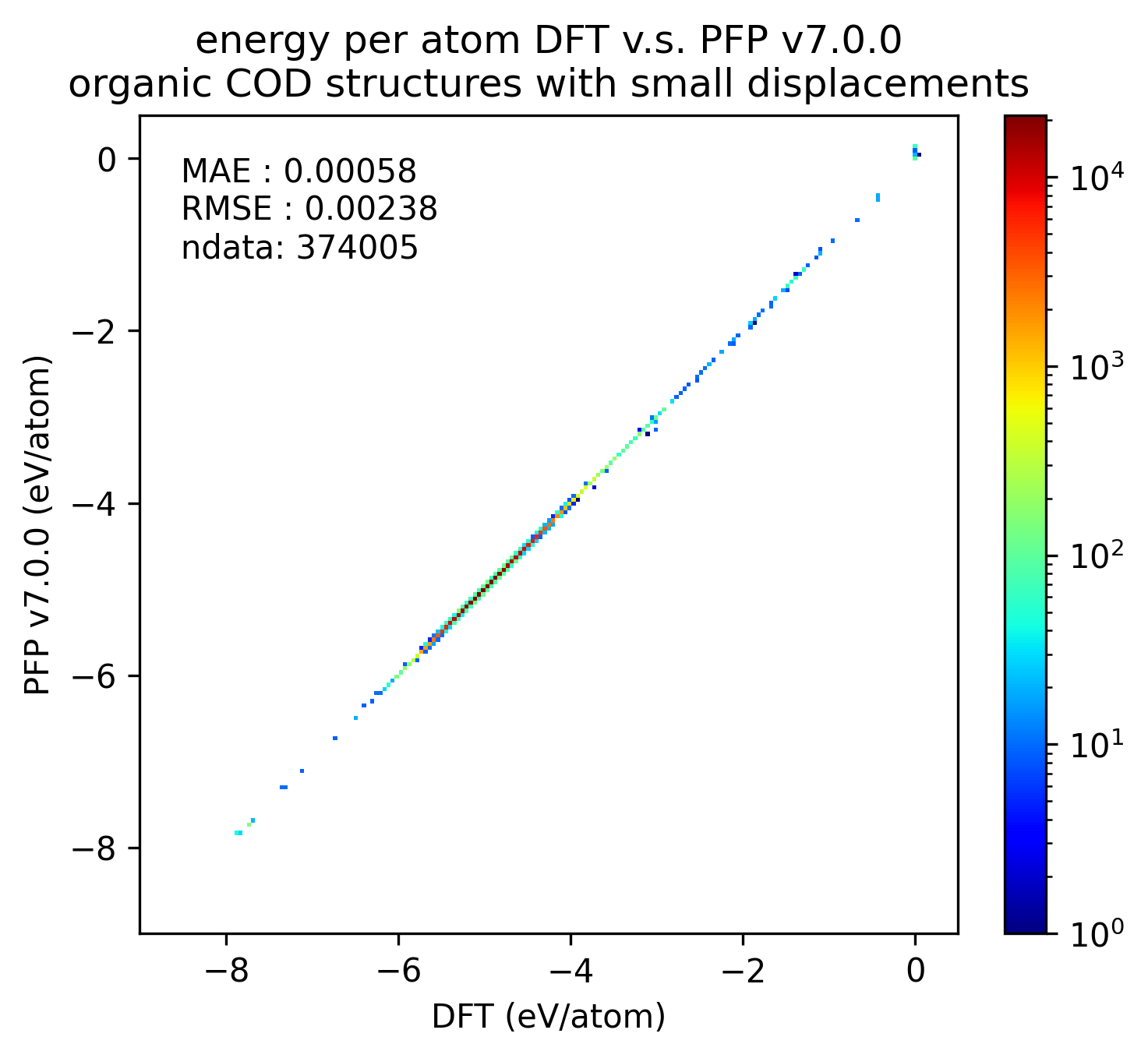 |
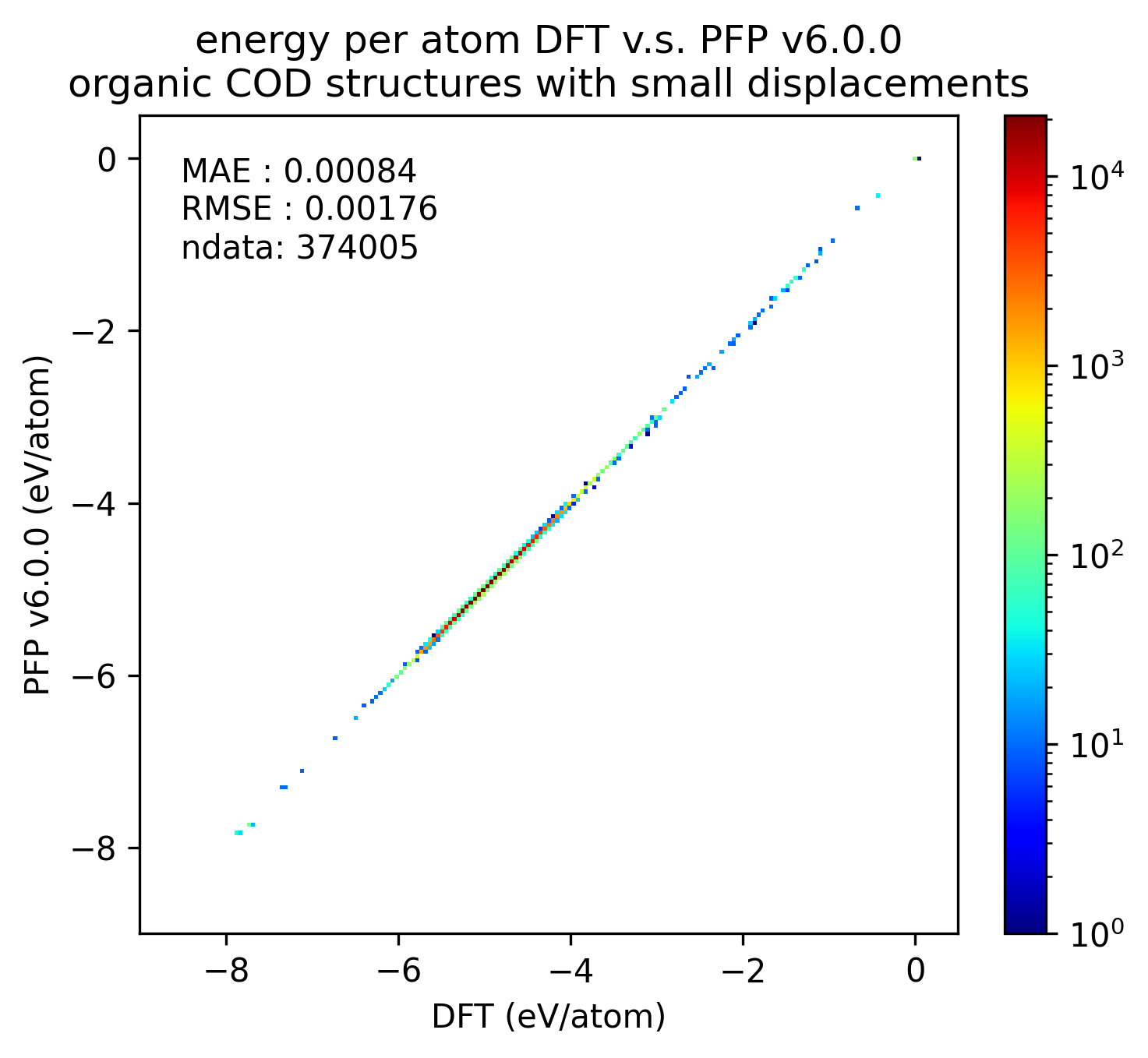 |
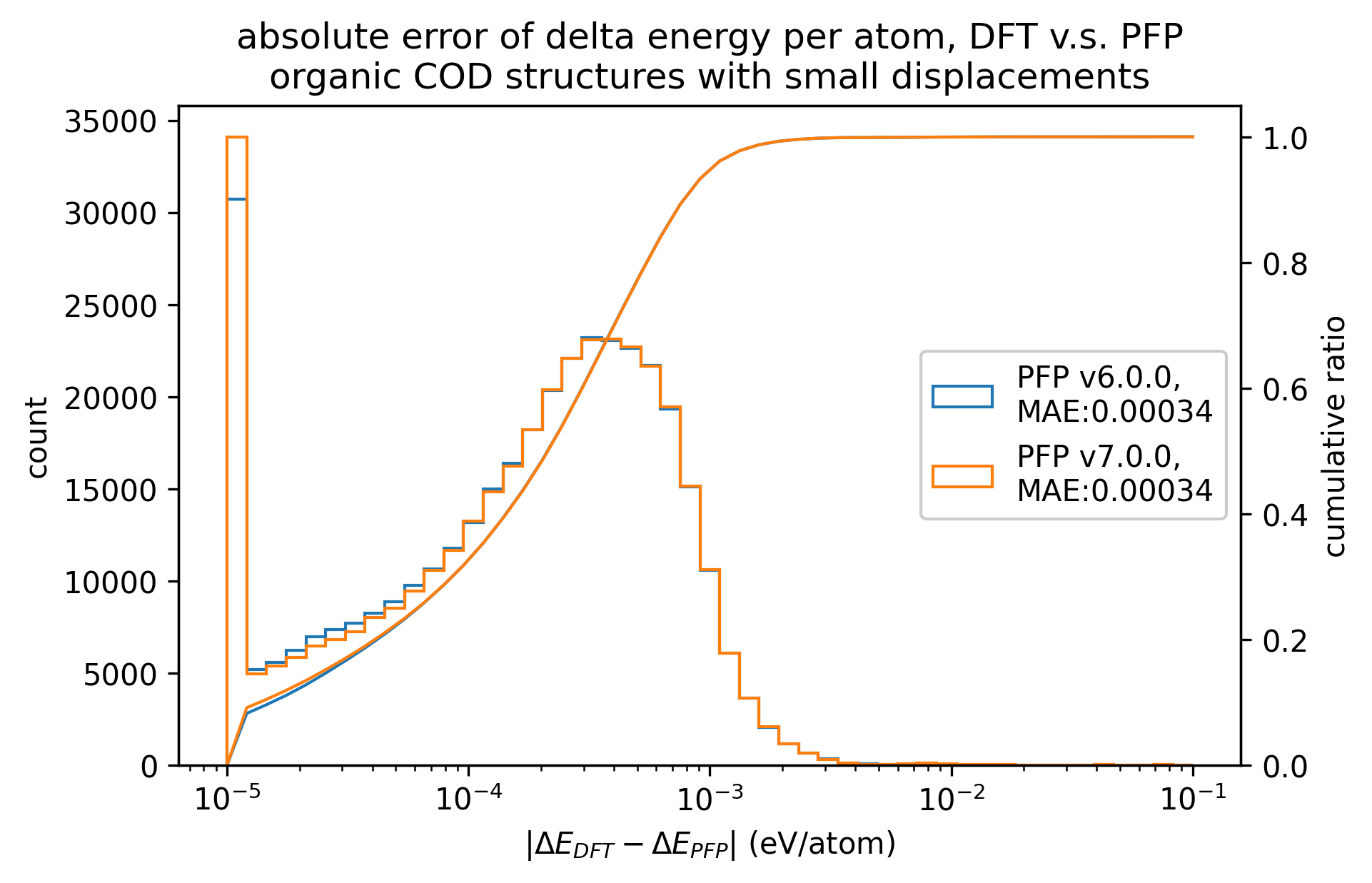 |
| 力 | 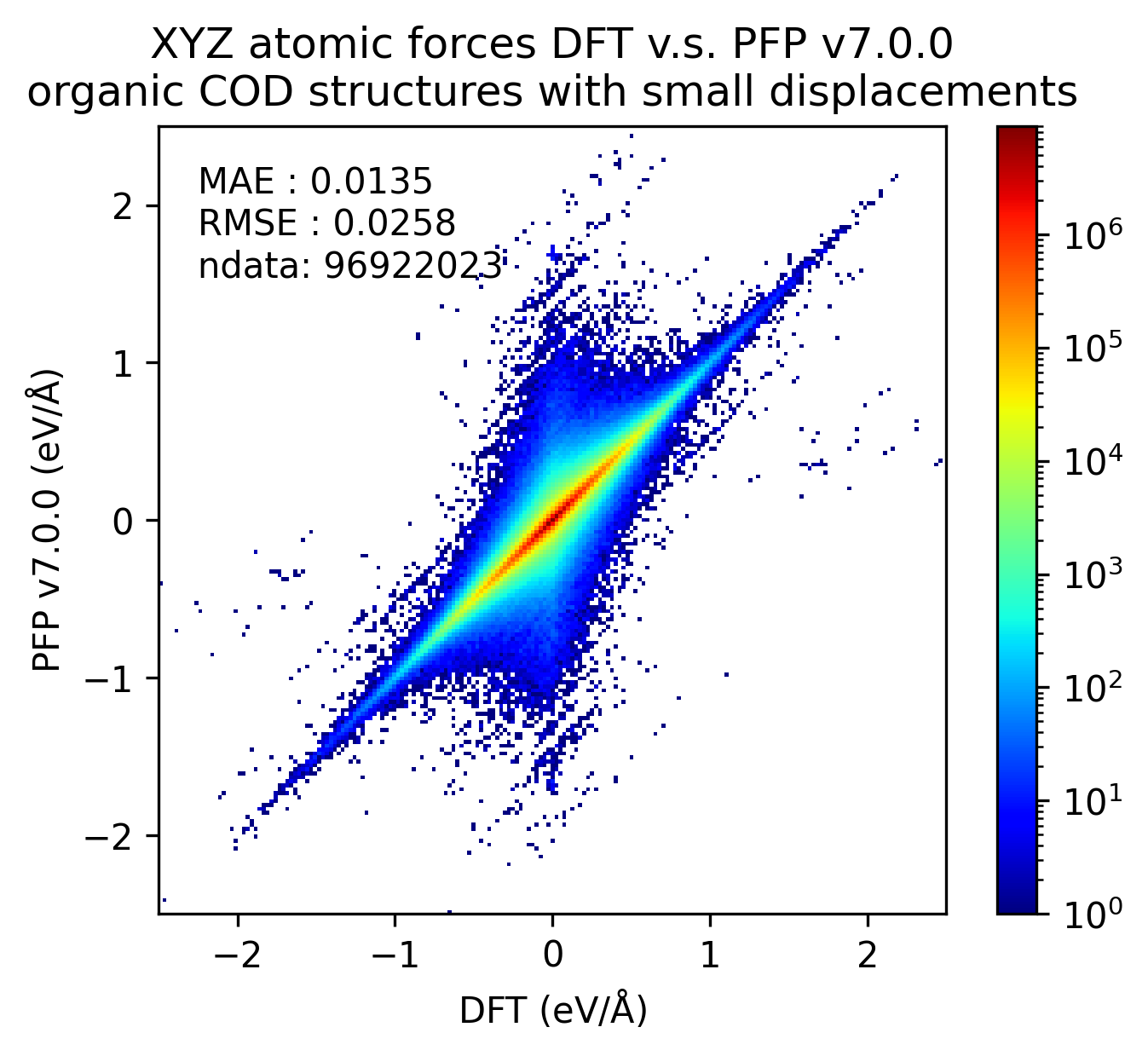 |
 |
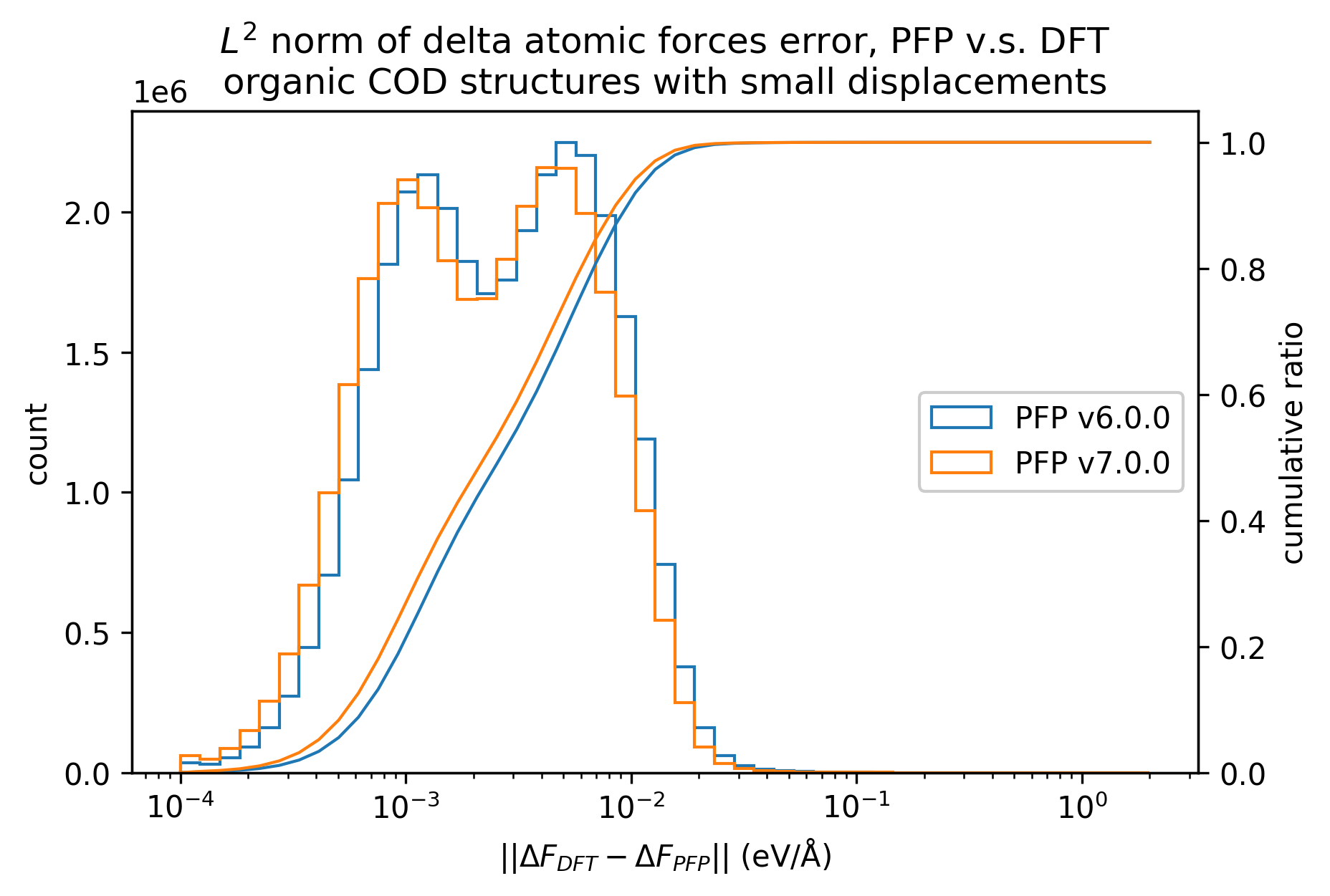 |
PFP v7.0.0では有機結晶に関して、エネルギー・力ともにMAEが改善していることがわかります。
微小変位を加える前後でのエネルギーと力の変化をDFTとPFPとで比較し、その誤差をヒストグラムとしてプロットしています。
| エネルギー | 力 |
|---|---|
|
|
微小変位を加える前後のエネルギーと力の変化は、v7.0.0とv6.0.0で同等です。
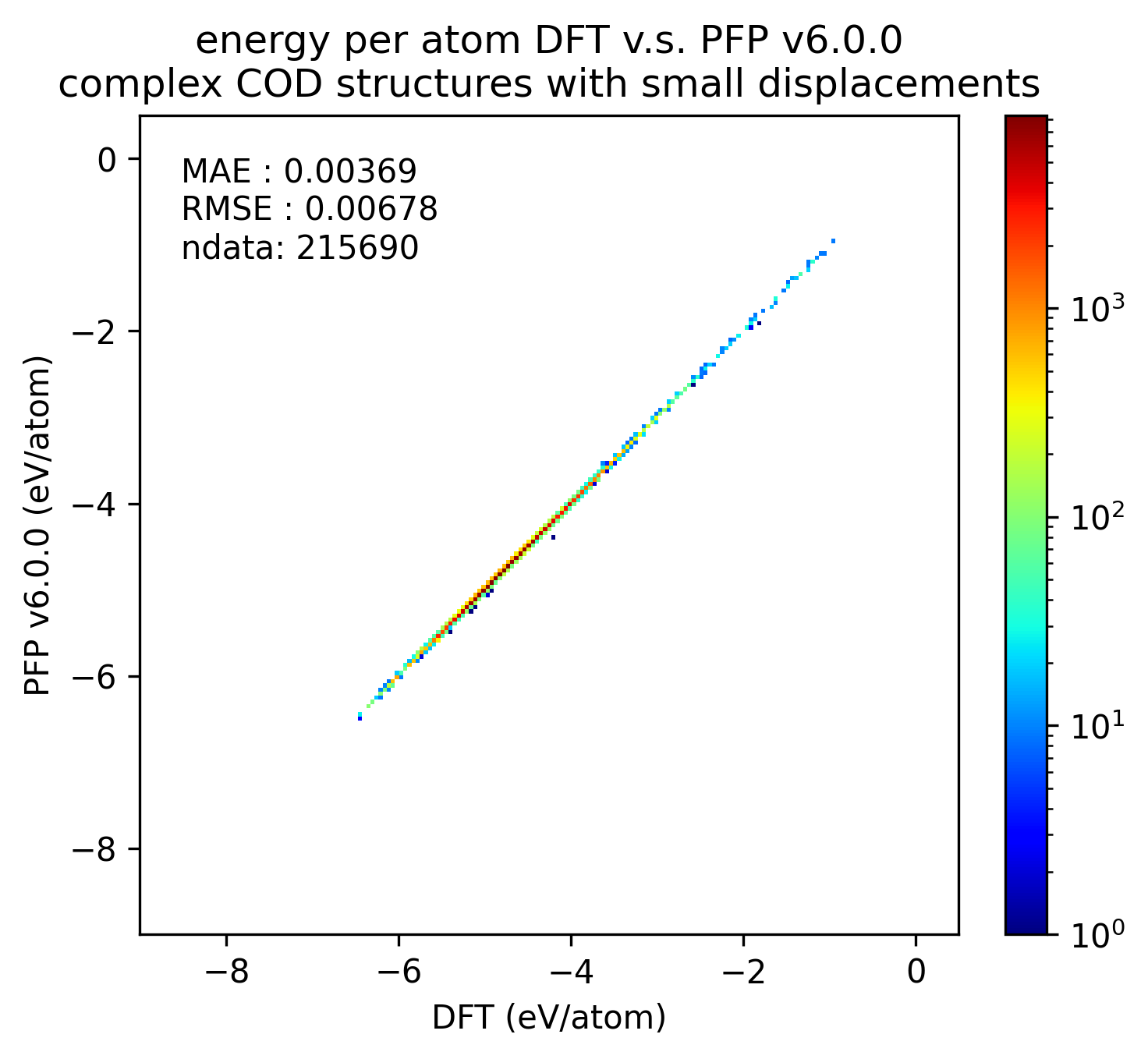
錯体結晶
以下の条件をすべて満たすものを錯体結晶とみなしました。
- H, C, N, O, P, S, F, Cl, Br, Iのうちの2元素以上を含むこと
- H, C, N, O, P, S, F, Cl, Br, I以外の元素を1元素以上含むこと
- H, C, N, O, P, S, F, Cl, Br, Iで原子数の8割以上を占めること
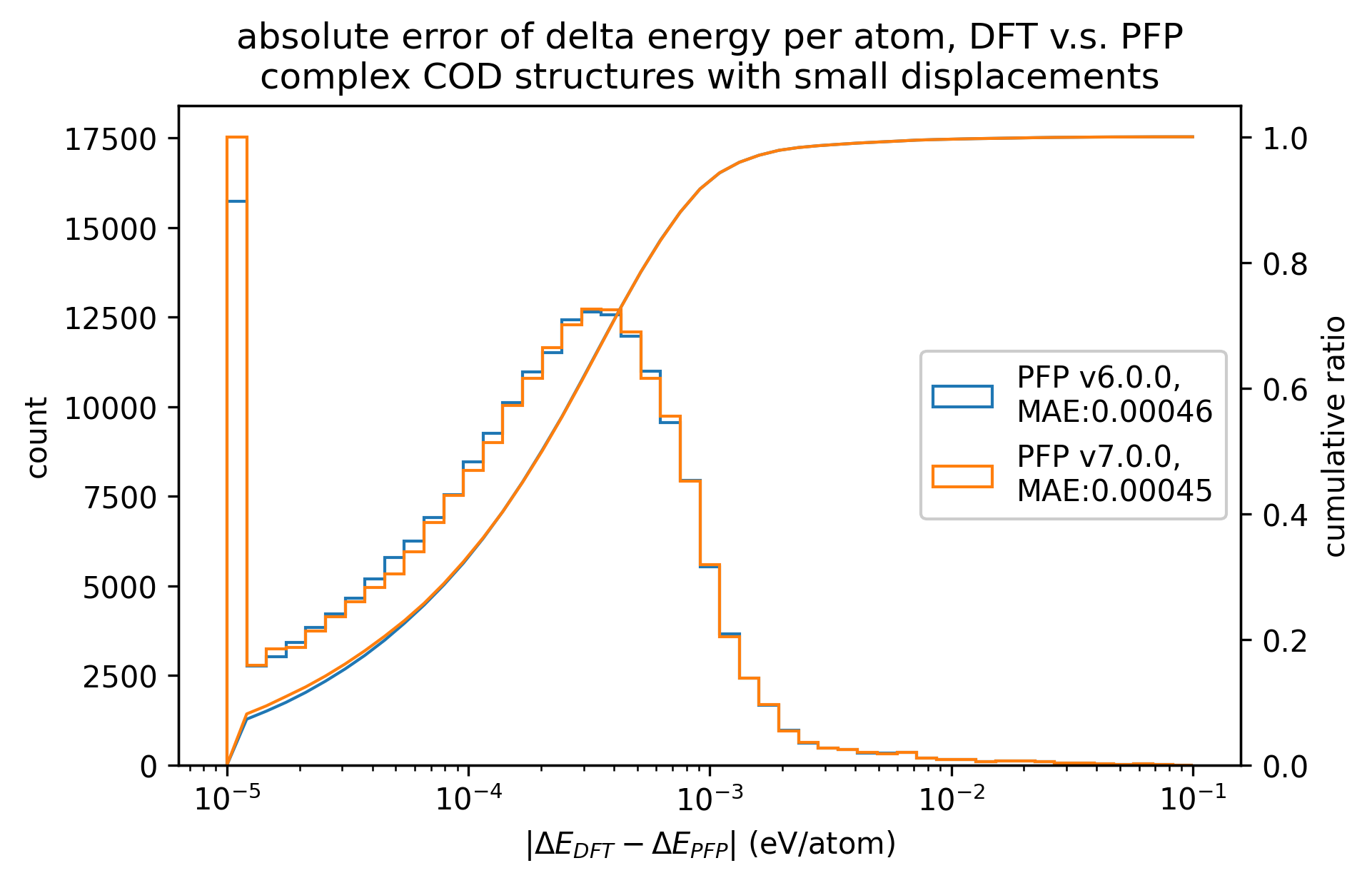
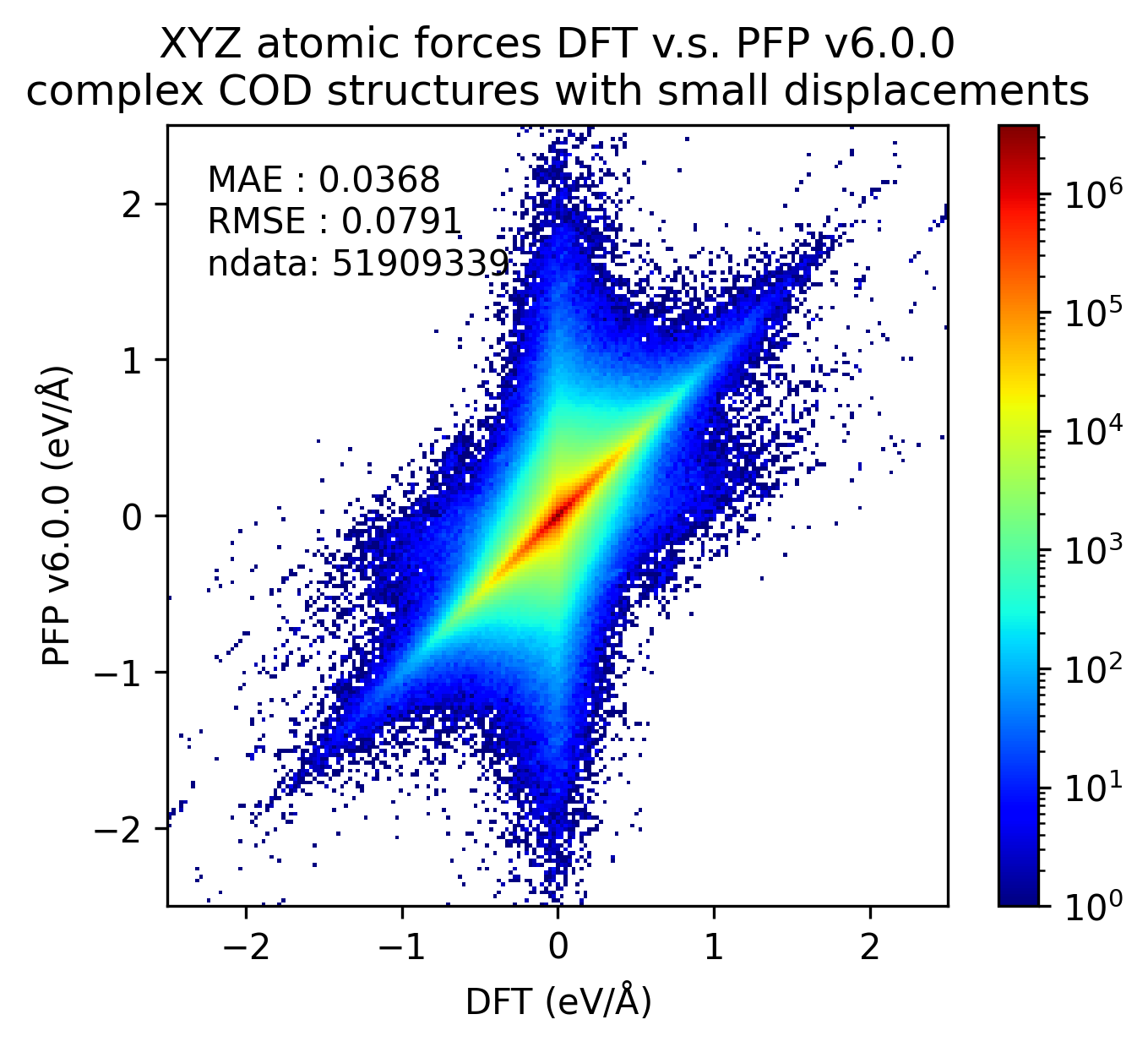
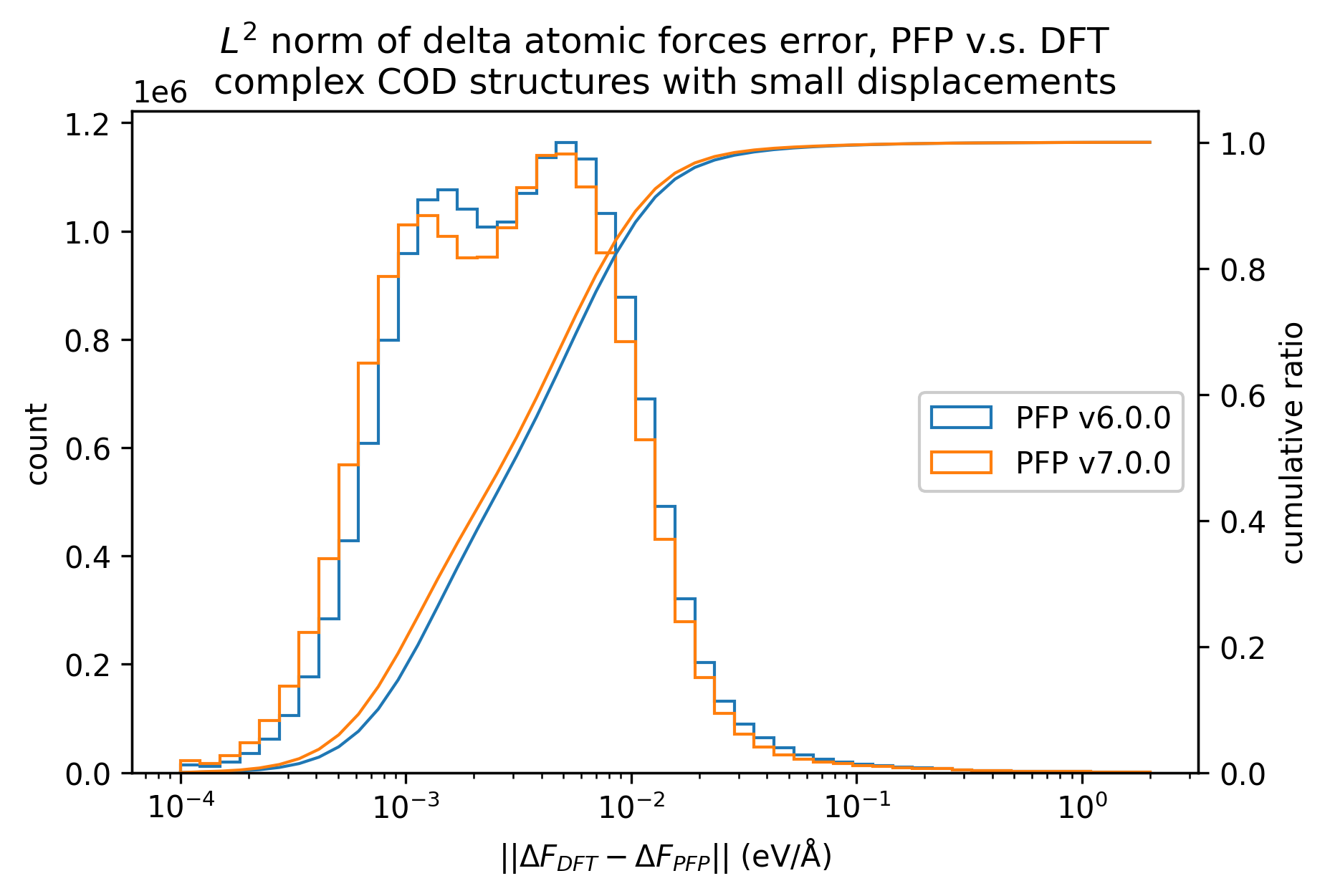
有機結晶と同様に、微小変位を加えた錯体結晶に対するエネルギー・力、および、錯体結晶に微小変位を加える前後でのエネルギー・力の変化を比較しました。
| v7.0.0 | v6.0.0 | histogram | |
|---|---|---|---|
| エネルギー | 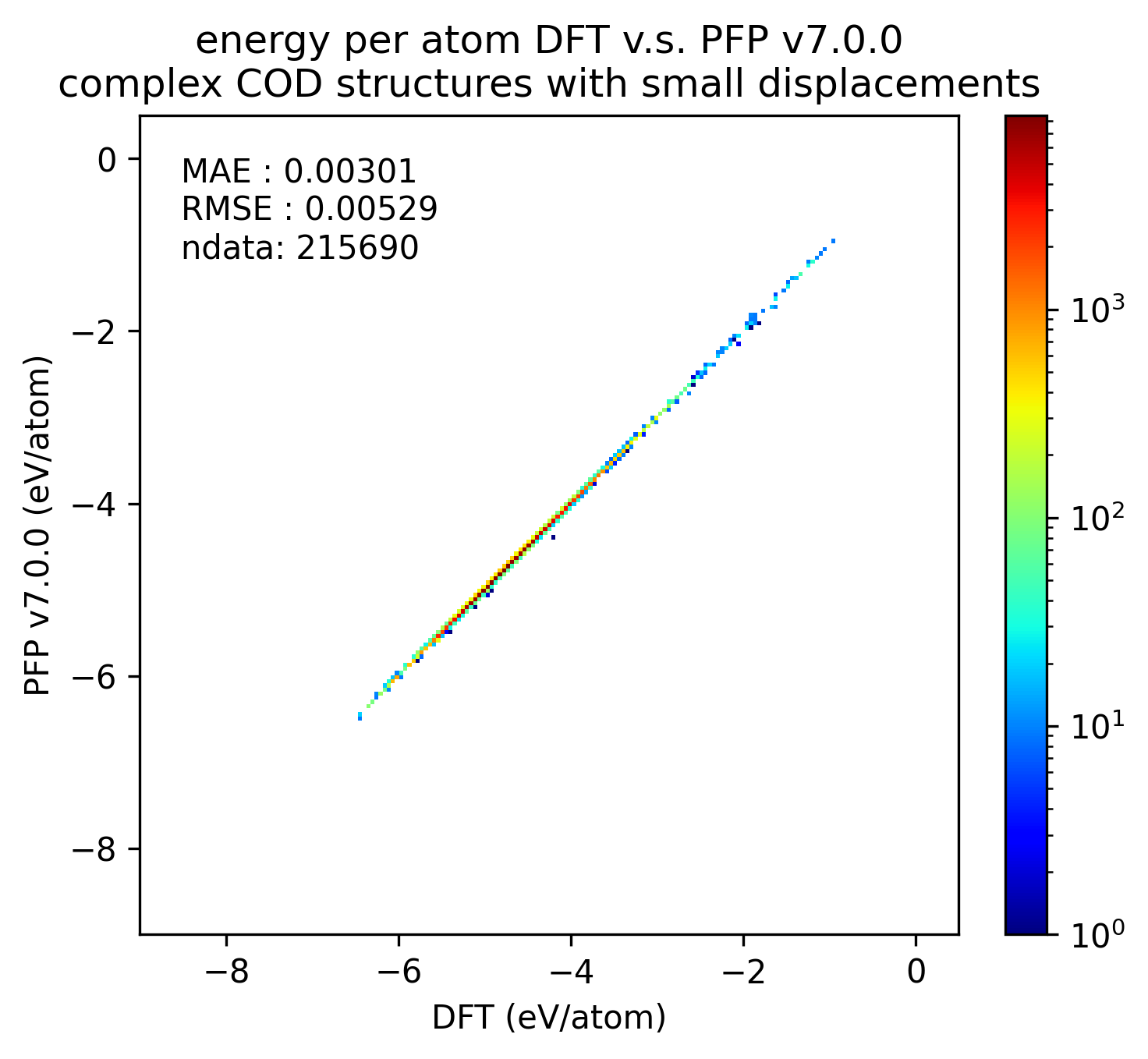 |
 |
 |
| 力 | 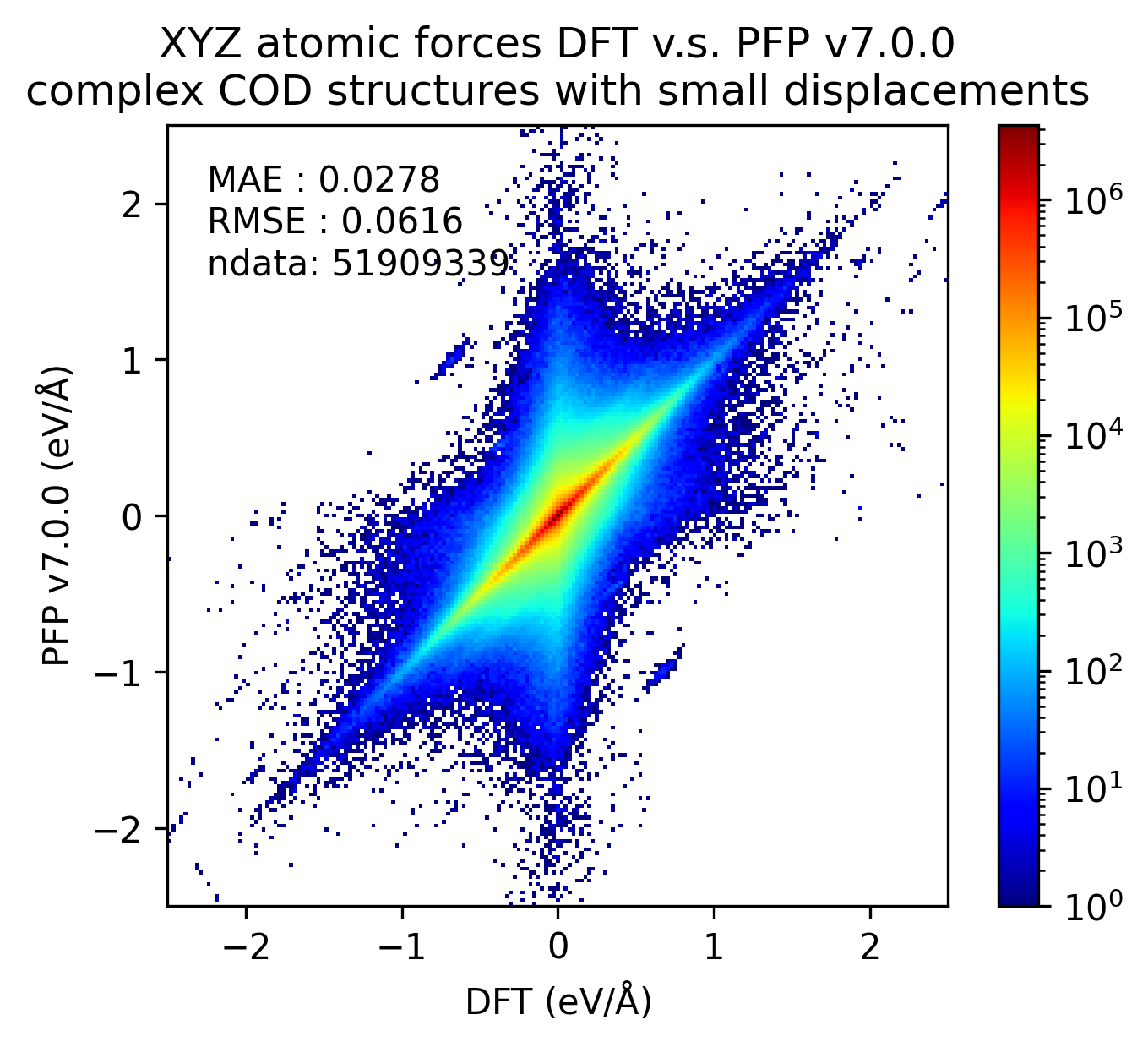 |
 |
 |
| エネルギー | 力 |
|---|---|
|
|
有機結晶に対する結果と同様、v7.0.0では錯体結晶に対するエネルギーと力の再現性が改善しています。
無機結晶
有機結晶と錯体結晶のいずれにも分類されない構造を、無機結晶とみなし、同様に比較しました。
| v7.0.0 | v6.0.0 | histogram | |
|---|---|---|---|
| エネルギー | 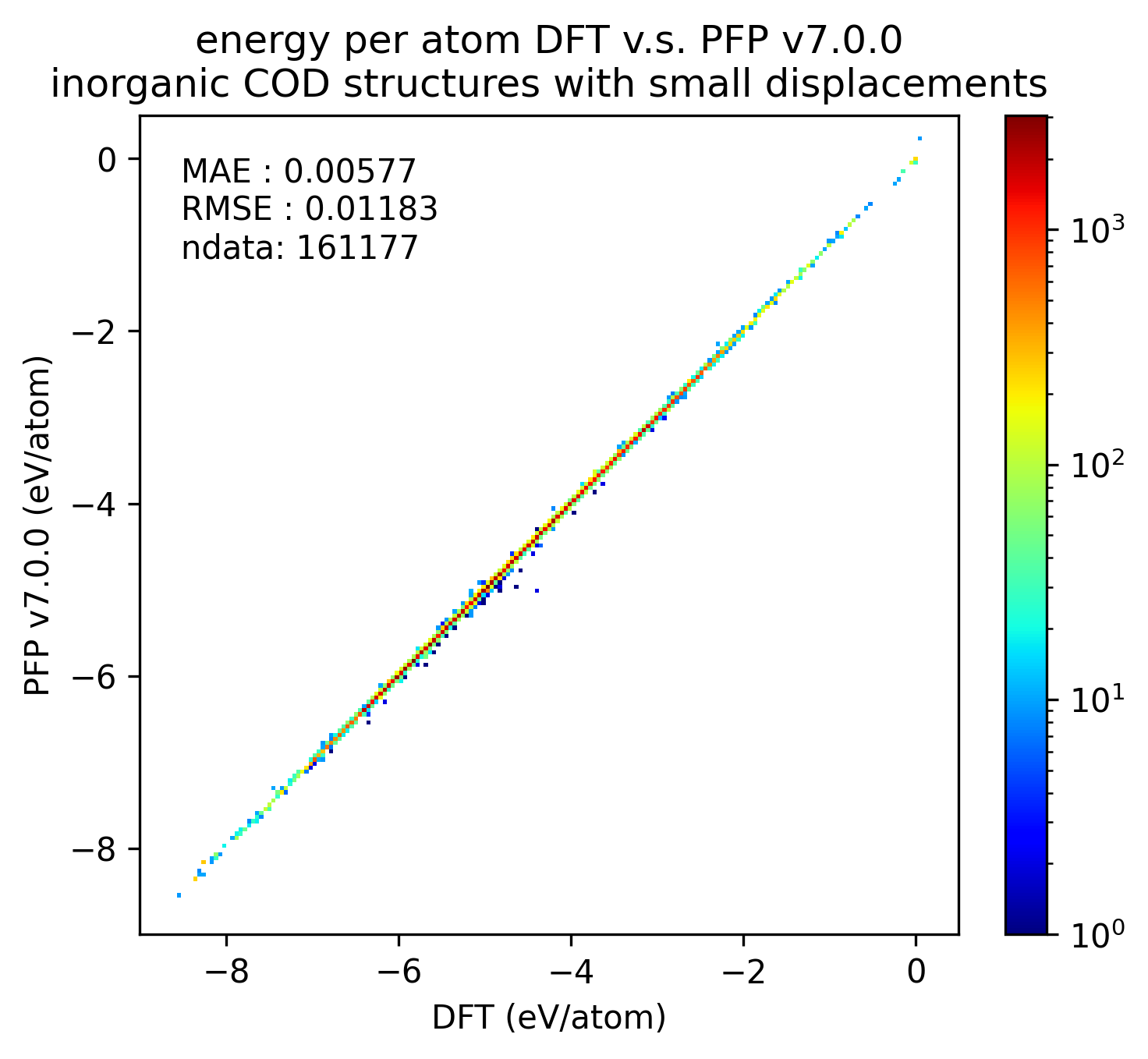 |
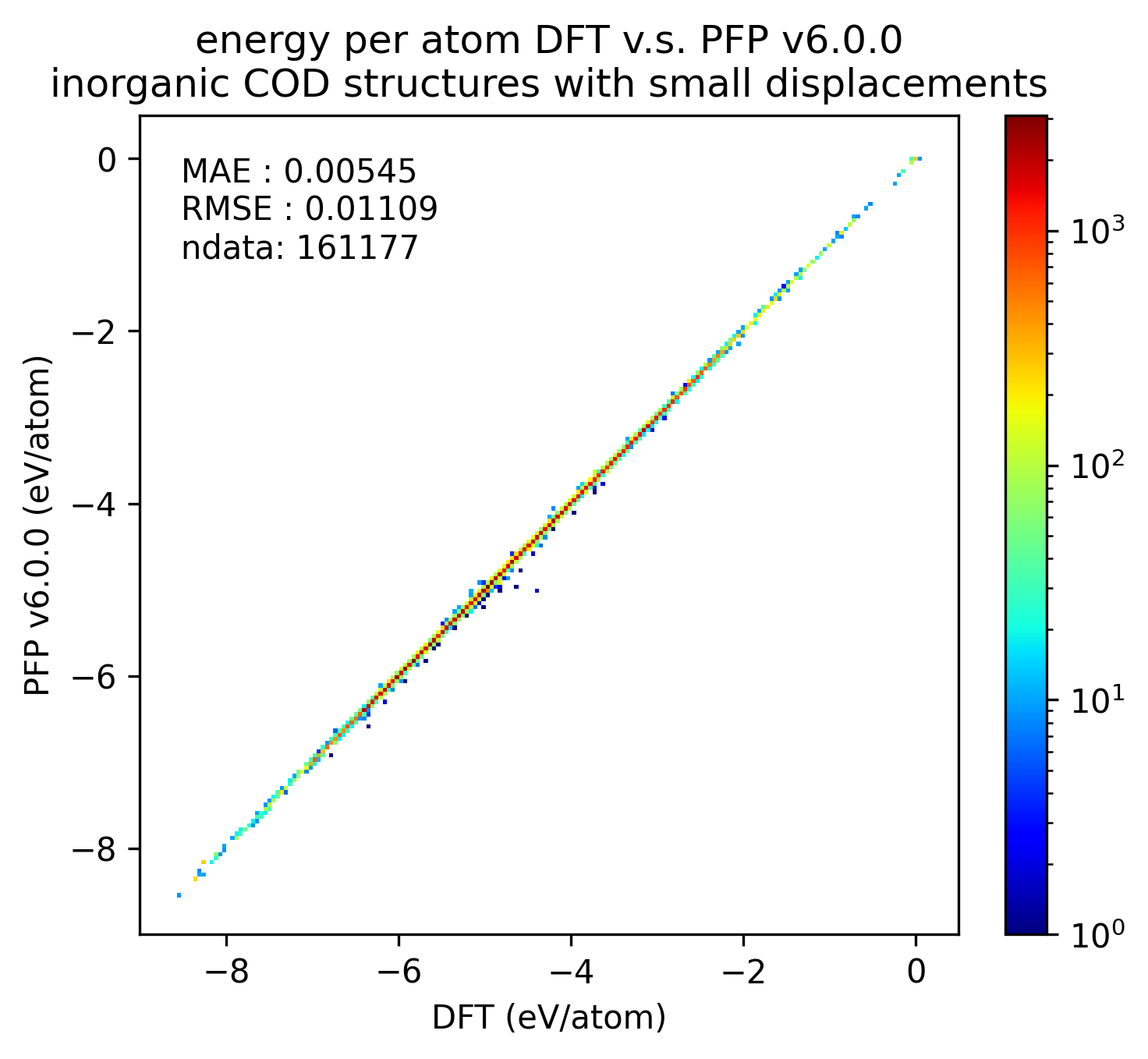 |
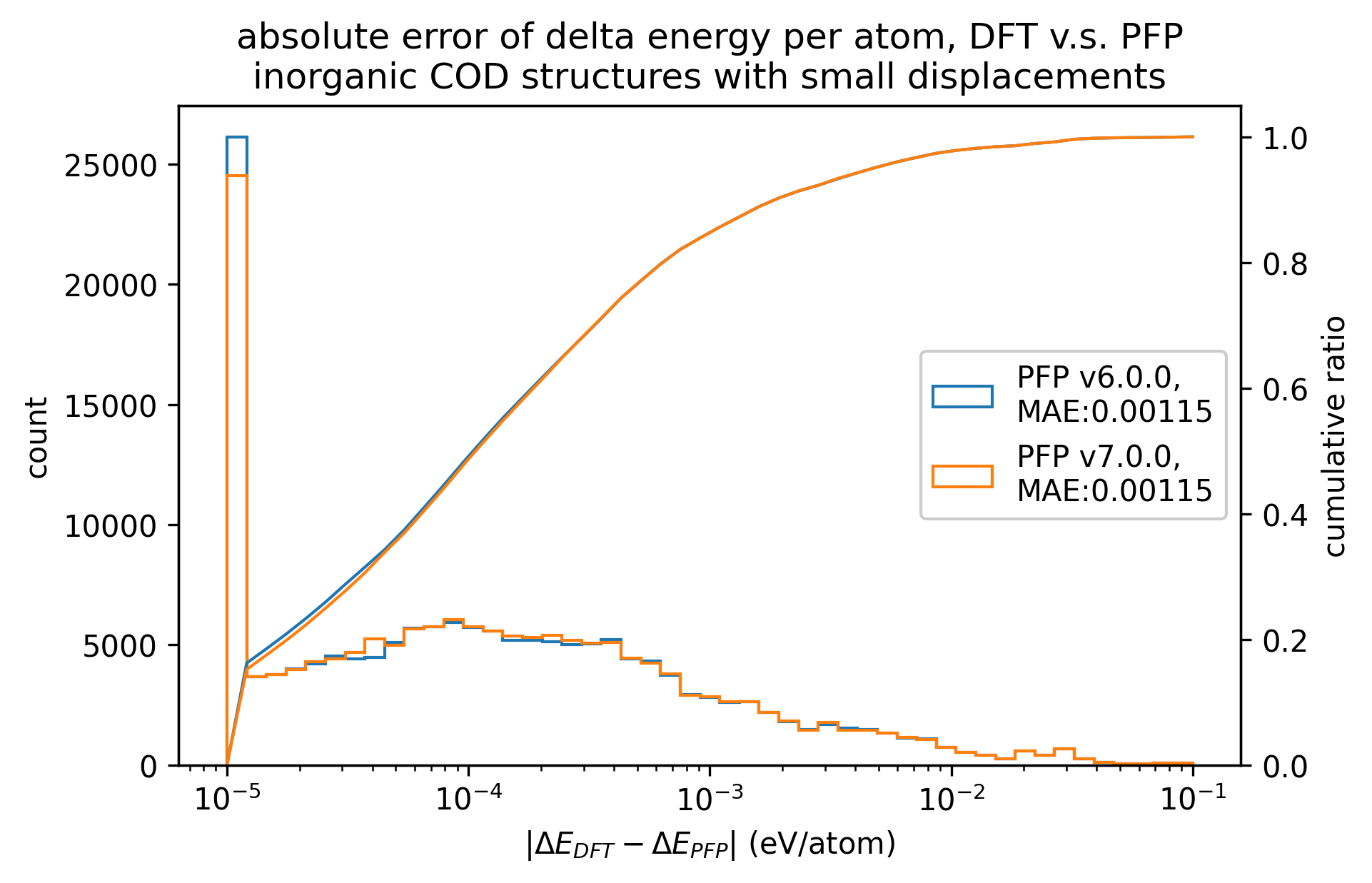 |
| 力 | 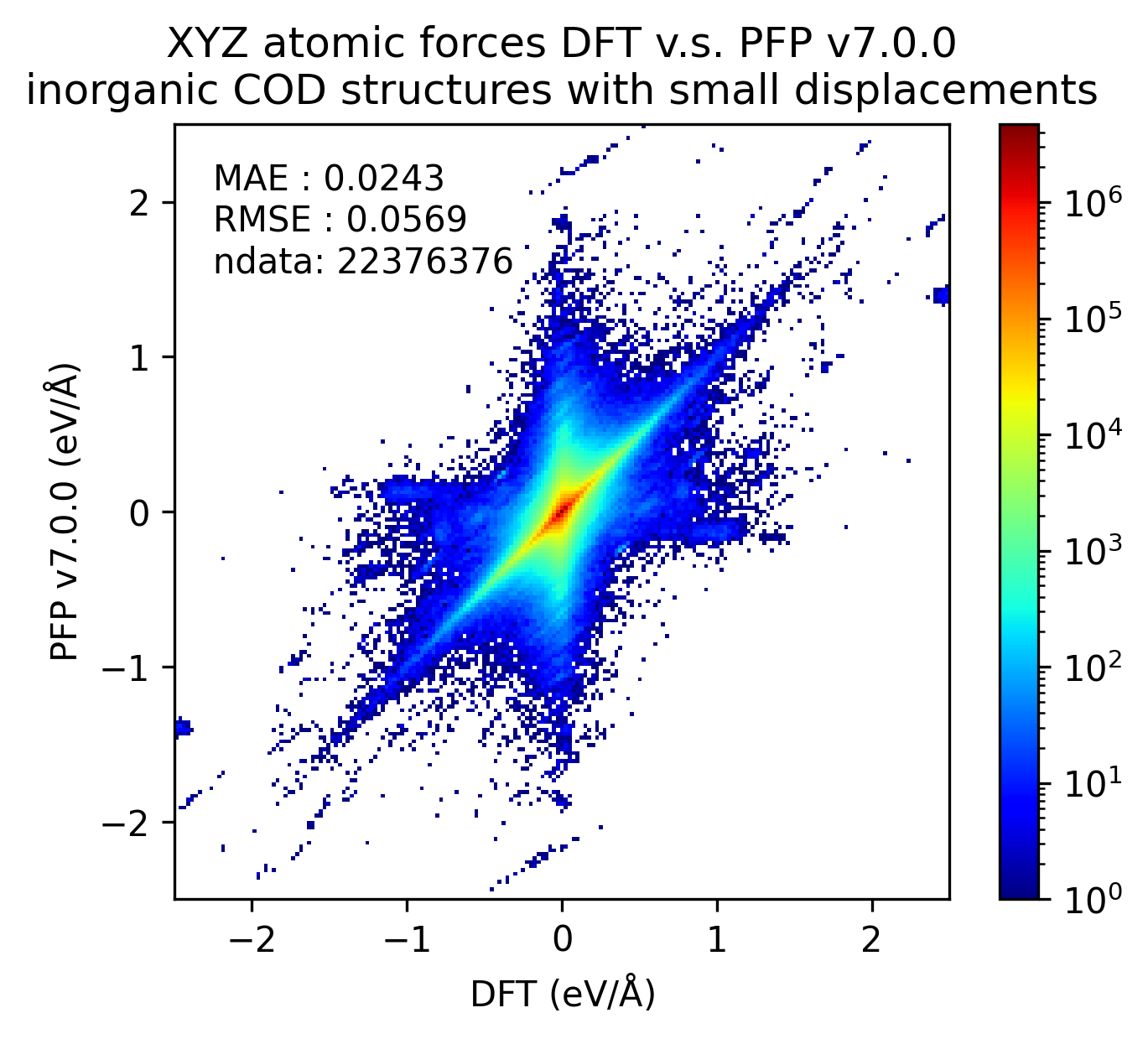 |
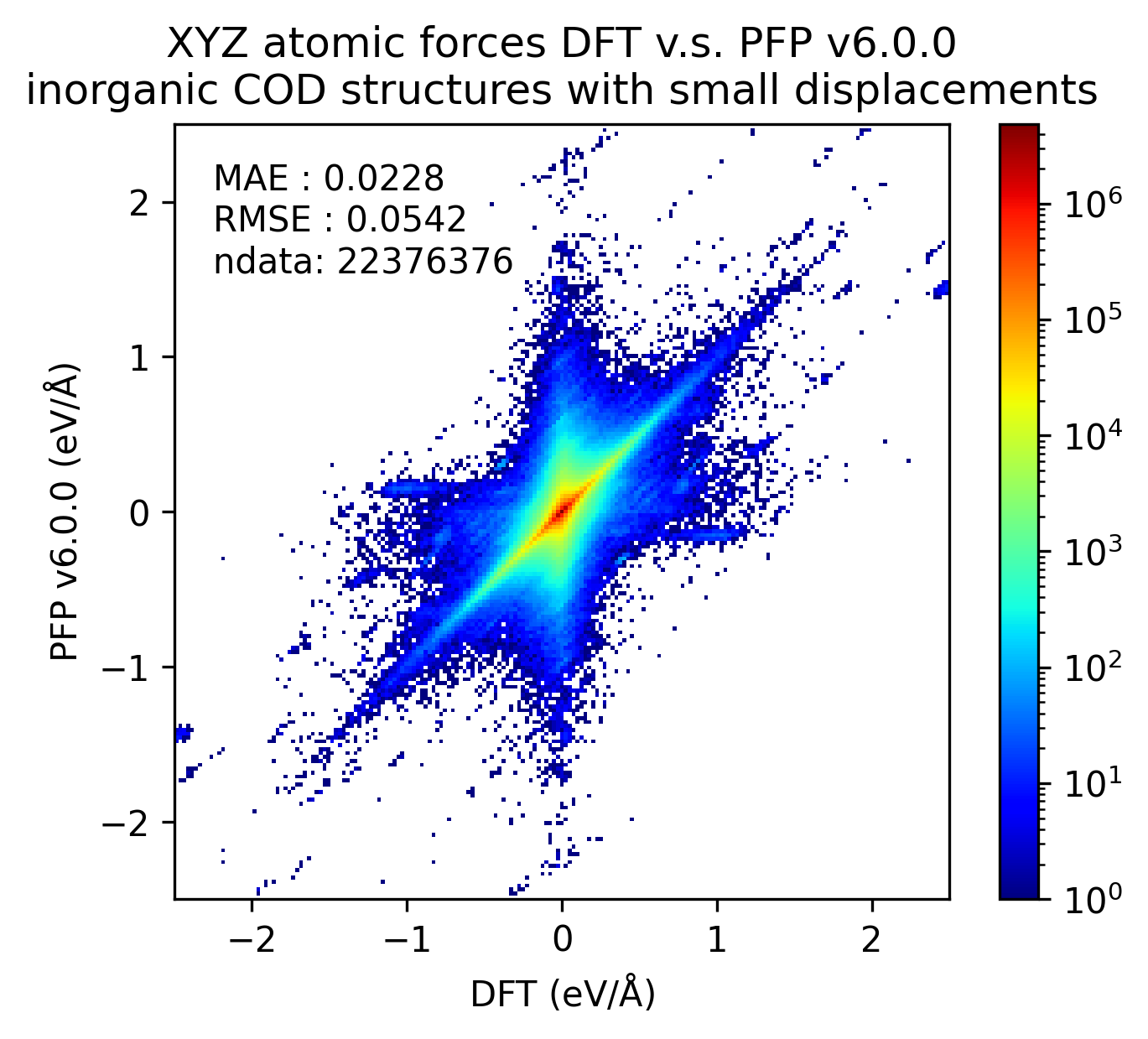 |
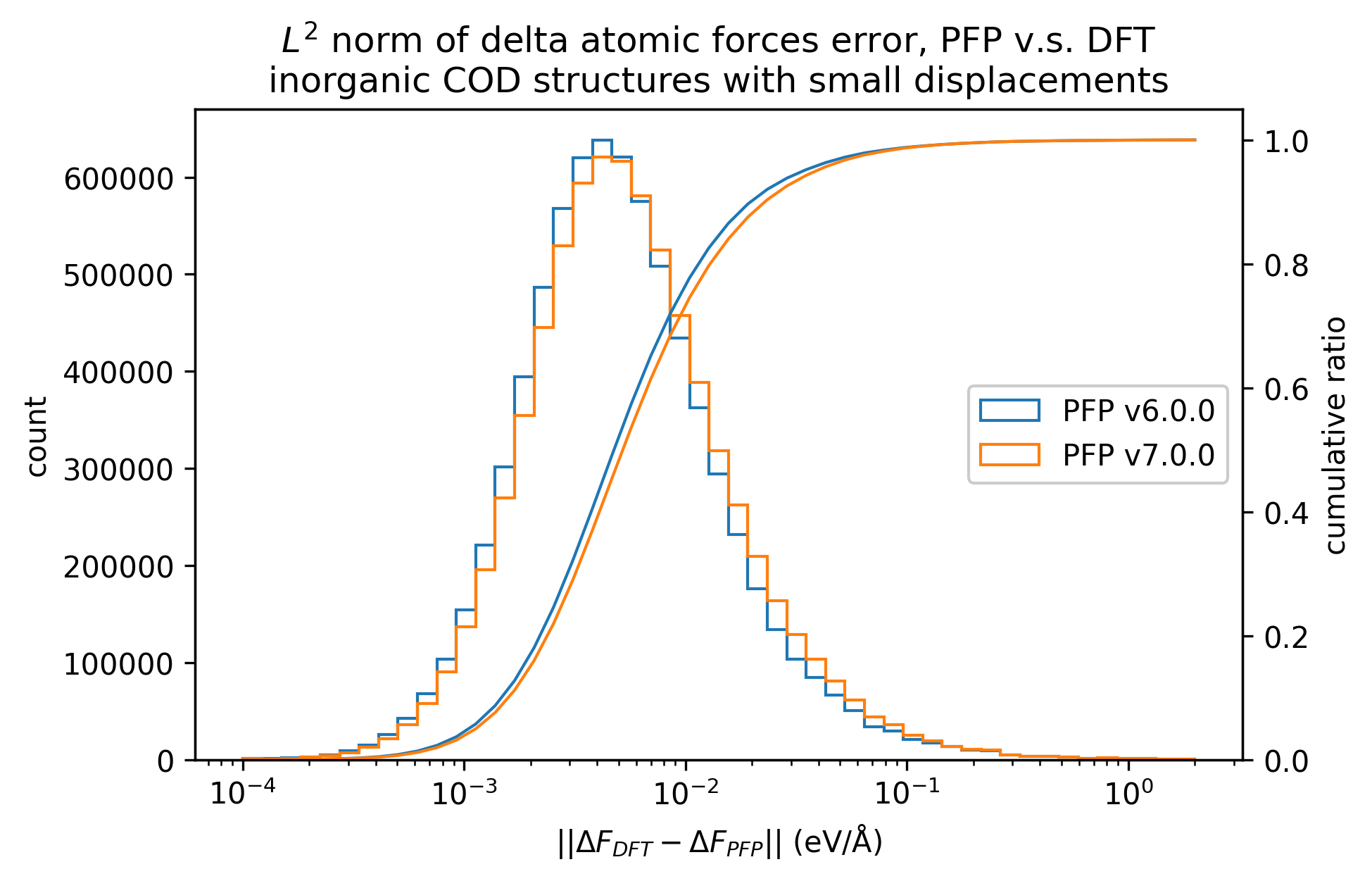 |
| エネルギー | 力 |
|---|---|
|
|
無機結晶に対するエネルギーと力の再現性はv7.0.0とv6.0.0とで概ね同等です。
体積と格子定数の再現性
DFTによる構造最適化後の構造を初期構造とし、PFPで格子定数を含む構造最適化を行い、体積の再現性をv7.0.0、v6.0.0、v5.0.0で比較しました。なお、前回のv6.0.0ではv5.0.0と比較して体積の再現性が低下していたため、v5.0.0も比較対象としています。
PFP v6.0.0でサポートされていた72元素のみを含む構造に対する結果です。
ase.Atomsであるatomsに対して、以下のコードを実行しています。
from ase.optimize import LBFGS
from ase.constraints import UnitCellFilter
ecf = UnitCellFilter(atoms)
opt = LBFGS(ecf)
opt.run(fmax=fmax, steps=2000)
fmaxの値として有機結晶と錯体結晶では0.03、無機結晶では0.01を用いています。
有機結晶と錯体結晶ではDFT-D3補正を用いられることが多く、本検証にあたってはDFT-D3補正を用いた場合の体積を有機結晶と錯体結晶に対して比較しています。
PFPではCRYSTAL_U0_PLUS_D3を指定しており、DFT計算ではCRYSTAL_U0での条件に加えてIVDW=12を指定することでDFT-D3補正を有効にしています。
無機結晶に対してはDFT-D3補正を用いていません。
| 相対体積 | 相対誤差 | |
|---|---|---|
| 有機結晶 | 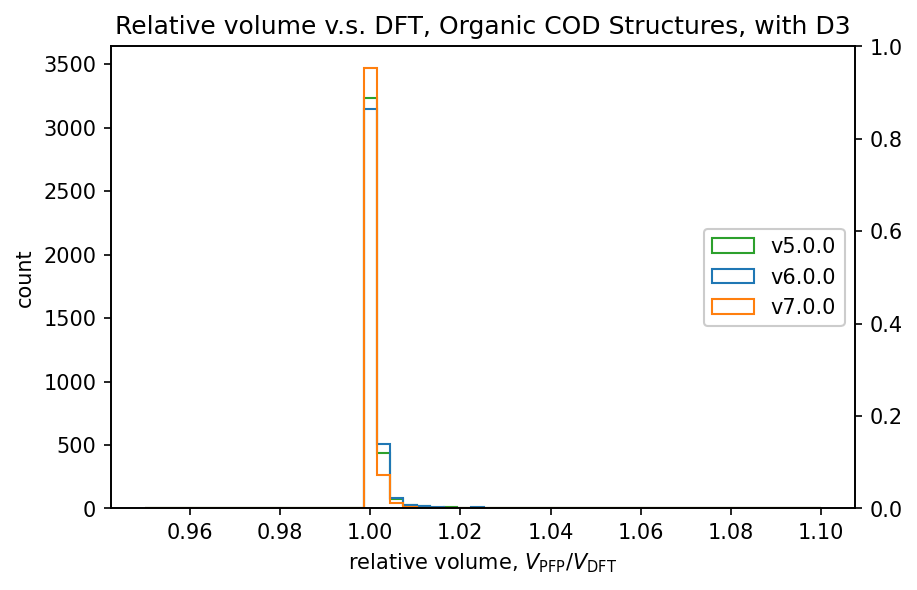 |
 |
| 錯体結晶 | 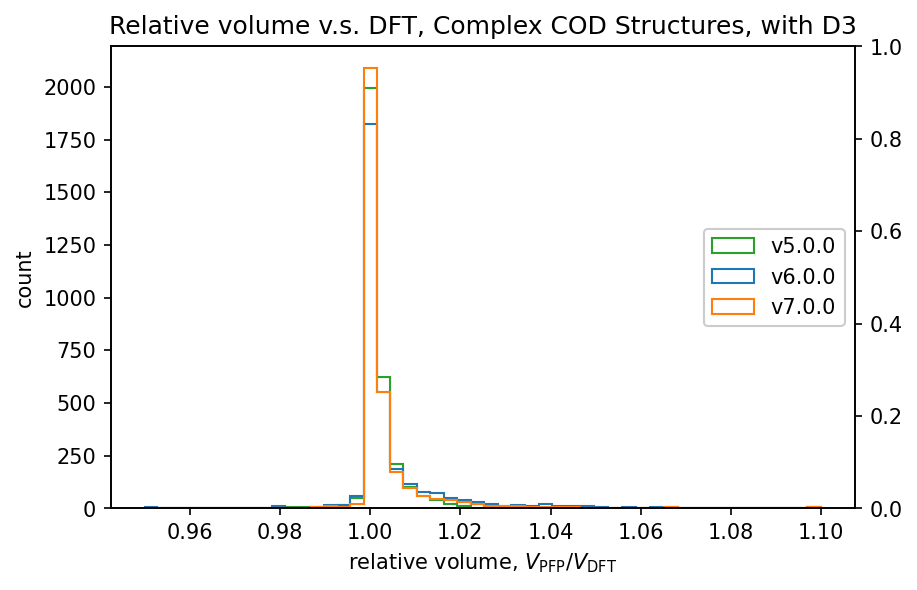 |
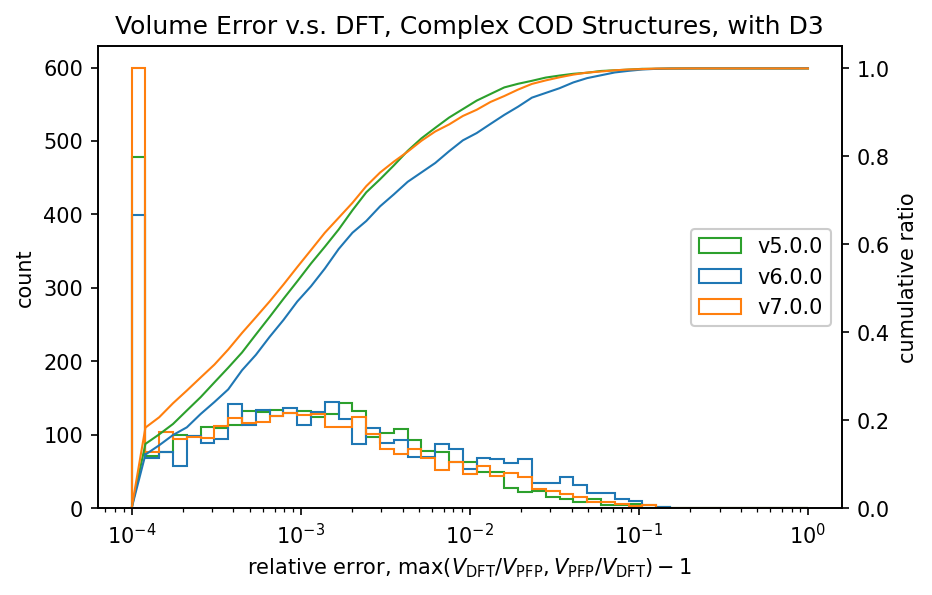 |
| 無機結晶 | 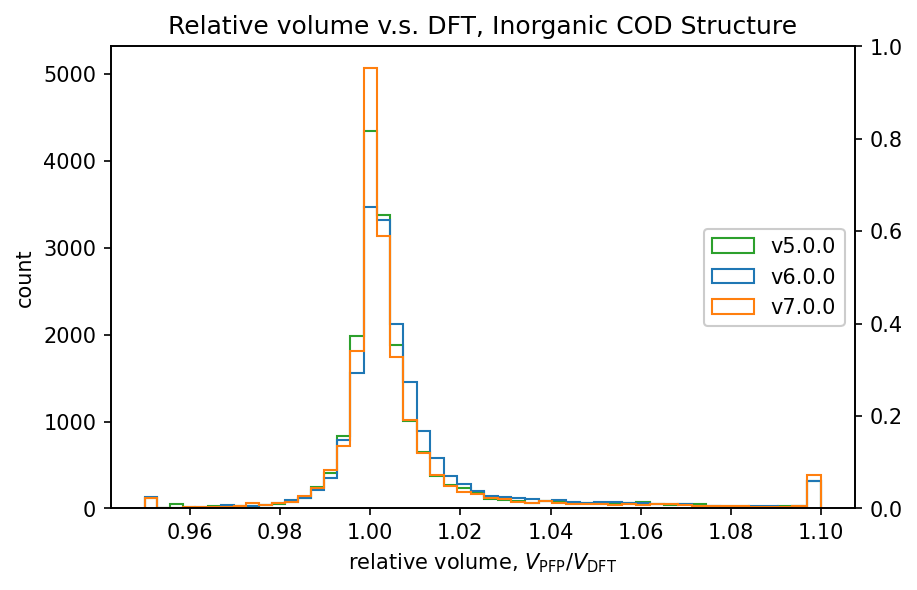 |
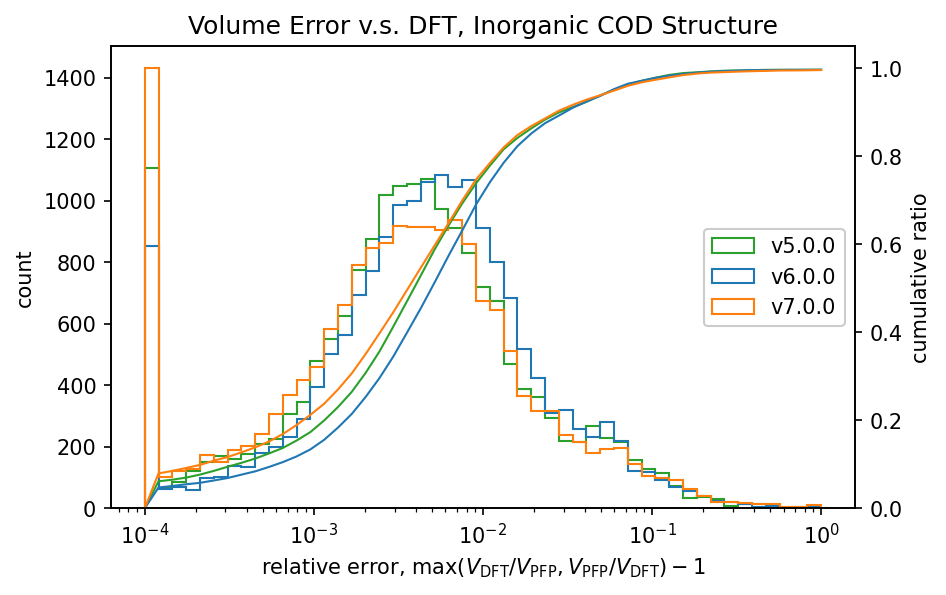 |
前回のv6.0.0では、v5.0.0と比較して体積の再現性が低下していました。
v7.0.0ではこれが改善し、特に有機結晶ではv5.0.0よりも高い再現性を示しています。
格子定数についての検証結果が以下です。
| 長さ | 角度 | |
|---|---|---|
| 有機結晶 | 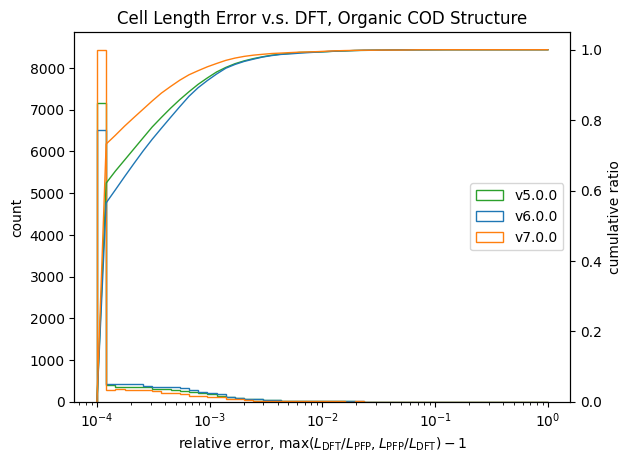 |
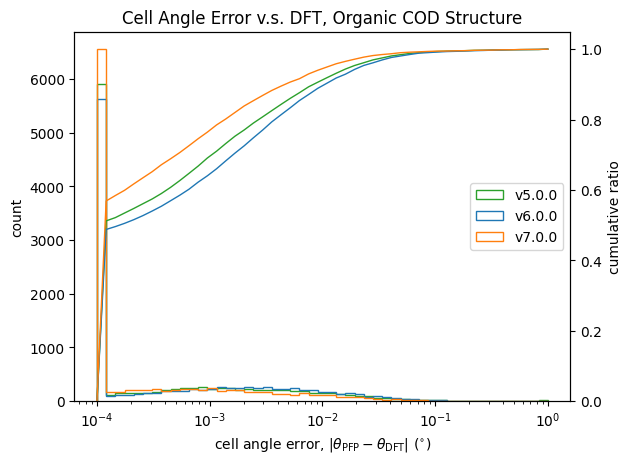 |
| 錯体結晶 | 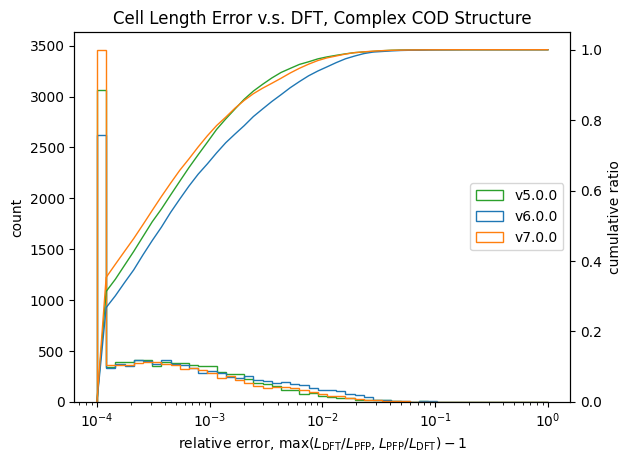 |
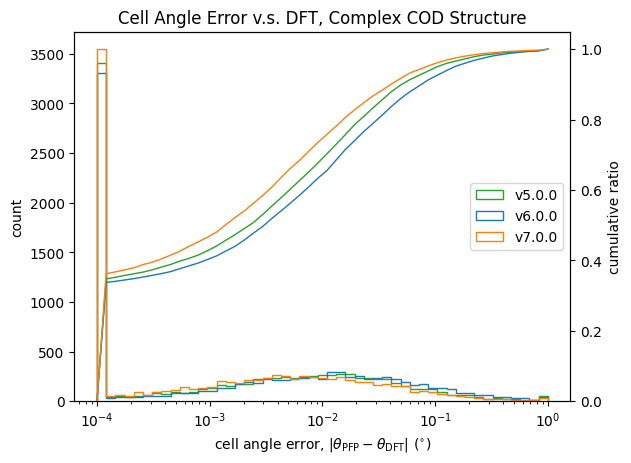 |
| 無機結晶 | 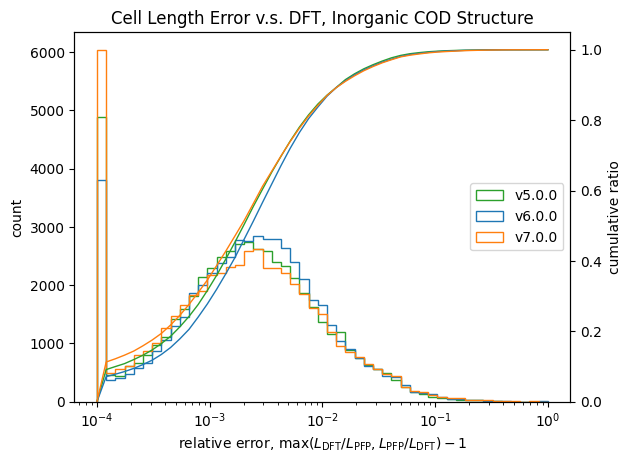 |
 |
こちらも体積と同様に改善が見られます。
COD構造によるベンチマークのまとめ
COD構造を有機結晶、錯体結晶、無機結晶の3つに分類してそれぞれ精度を比較しました。
PFP v7.0.0は有機結晶および錯体結晶について、エネルギーと力の再現性が改善しています。
また体積に関しても再現性の向上が見られました。
液体の密度の再現性
25種類の分子を対象として液体構造の密度の実験値との比較を行います。
計算条件
計算に使用した25種類の分子のリストは以下の通りです。
ethylene glycol, acetic acid, benzene, n-octane, DMSO, water, pyridine, toluene, Et2O, IPA, chloroform, DMF, acetonitrile, methane sulfonic acid, trifluoroethanol, 1-methylnaphthalene, GBL, xylene, PC, THF, n-hexane, EtOAc, perfluorooctane, HMPA, dioxane
液体の構造のため、有限温度での分子動力学(MD)計算を行います。それぞれの分子について、以下の手順でシミュレーションを行いました。
- 系の原子数が400以上になるように分子を複製
- 300 KのNVTアンサンブルで1 psのMD
- 300 K、1気圧のNPTアンサンブルで100 psのMD
- NPTアンサンブルのtrajectoryのうち最後の20 psについて平均密度を計算
今回はすべての分子について上記の計算条件で計算しました。
細かい計算条件は以下の通りです。
- PFPの計算モードは
CRYSTAL_PLUS_D3(D3補正あり) - MDのtimestepは全体を通して1 fs
- NPTアンサンブルでの体積変化は等方変形のみ許可
また今回のPFP Validationからpfactorなどの計算条件を変更しています。
密度の比較
計算された密度と実験値との比較を以下に示します。参考までに、PFP v6.0.0で同じ計算を行ったものを同様に示しています。
| PFP v7.0.0 | PFP v6.0.0 |
|---|---|
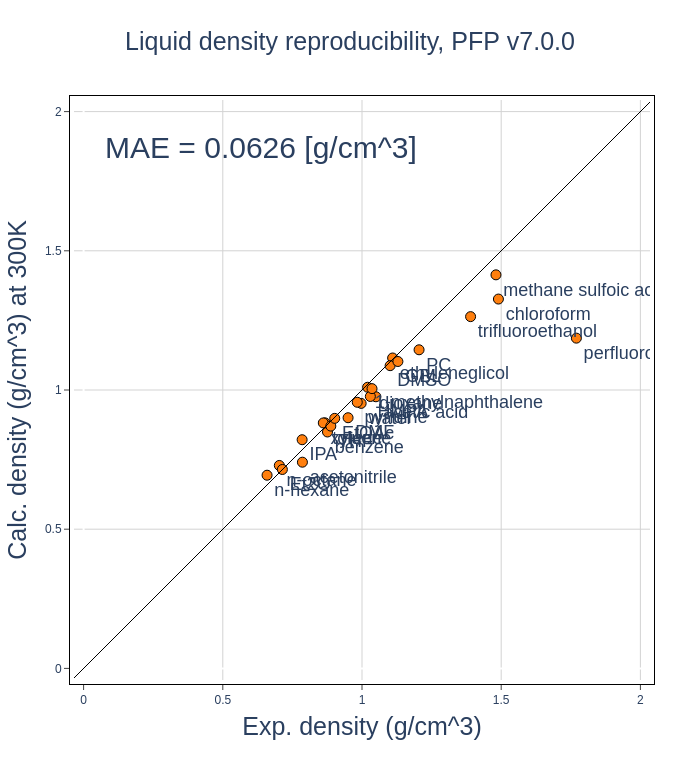 |
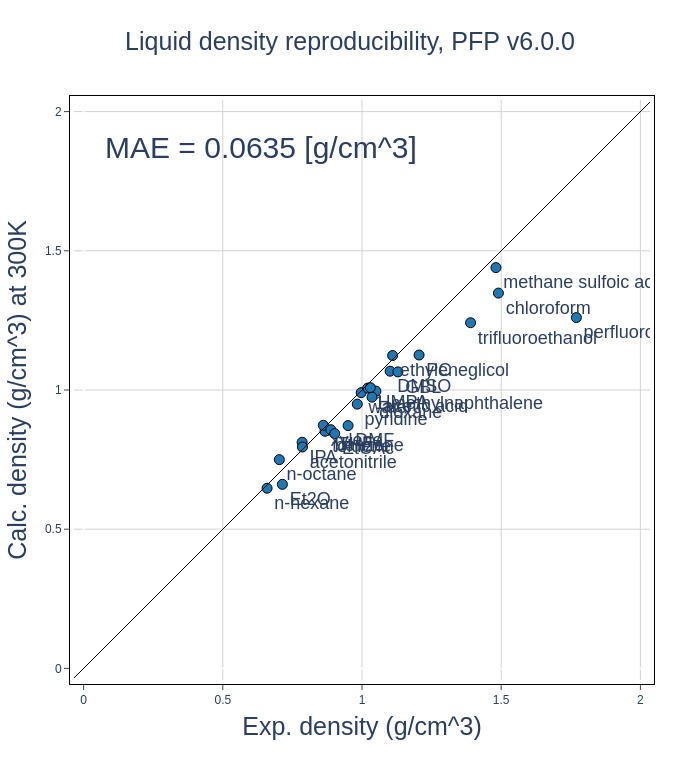 |
v7.0.0とv6.0.0での再現性に変化はなく、どちらもperfluorooctaneに関する再現性が特異的に低いことが分かります。
perfluorooctaneは比較的長鎖のパーフルオロアルカンです。
何らかの要因により、これらの分子に対してv5.0.0以降のPFPの再現性が特異的に低いと考えており、その要因を調査中です。
相図の再現性によるベンチマーク
ここでは相図の再現性を確認するため、絶対零度における組成をふった相図(状態図)を描画し、DFT計算結果との比較を行いました。
ベンチマークデータセット
結晶構造のデータベースとして、Materials Projectに登録されている結晶構造を使いました。DFTの計算条件の違いを吸収するため、PFPデータセットの計算条件(CRYSTAL_U0に相当するもの)により再計算を行いました。再計算では格子定数を含めた構造最適化を行っており、収束条件はforceが0.005 eV/Å以下としました。なお、今回はベンチマークの問題設定の都合上、これらの初期構造の多くはPFPの学習用データセットにも含まれています。
構造最適化計算
これらの結晶構造を初期構造として、PFPのCRYSTAL_U0モードで構造最適化計算を行い、PFPにおける緩和構造を得ました。PFPの構造最適化計算にはASEのLBFGS法を使い、fmax=0.01, alpha=300, steps=4000としています。PFPの構造最適化計算においても、格子定数を含めた構造最適化を行っています。
最終的に得られた結晶構造は95347個です。
相図の再現性
Energy above hull
相図は、元素の組成と形成エネルギーの空間に様々な結晶構造を配置した図であり、特に安定に存在しうる結晶構造群からなる凸包(convex hull)として表現されます。用語の区別として、ここでは構造最適化計算で得られた局所安定な構造を緩和構造、相図を描いたときに凸包を構成する構造を安定構造と呼ぶことにします。ここでは相図の再現性の指標として、相図を描いたときの安定構造以外の結晶構造のエネルギー(energy above hull)の予測精度に着目しました。これは、相図における凸包が正しく描けていることに加え、類似の組成を持つ結晶構造間のエネルギーの差が正しく表現できているかどうかを測る指標になります。
具体的な手順は以下のようになります。まず結晶構造のデータに対し、1, 2, 3元系の相図として考えられるすべての元素の組み合わせにおいて相図を描くことを考えます。これをDFT計算で得られたエネルギーとPFPにおける構造最適化計算後のエネルギーの2パターンについてそれぞれ行います。得られたすべての相図に対して、その中のすべての構造についてenergy above hullを計算します。DFT計算の相図でenergy above hullが0より大きい構造(=安定構造以外の構造)を抽出し、DFTとPFPの2つの相図でenergy above hullを比較します。
得られた結果を以下に示します。比較のためにPFP v6.0.0の結果も示しています。x, y軸がそれぞれlogスケールになっていることに注意してください。PFP v7.0.0はv6.0.0と比較して、結晶構造のエネルギーの予測精度がわずかに下がっていることが分かります。
| PFP v7.0.0 | PFP v6.0.0 |
|---|---|
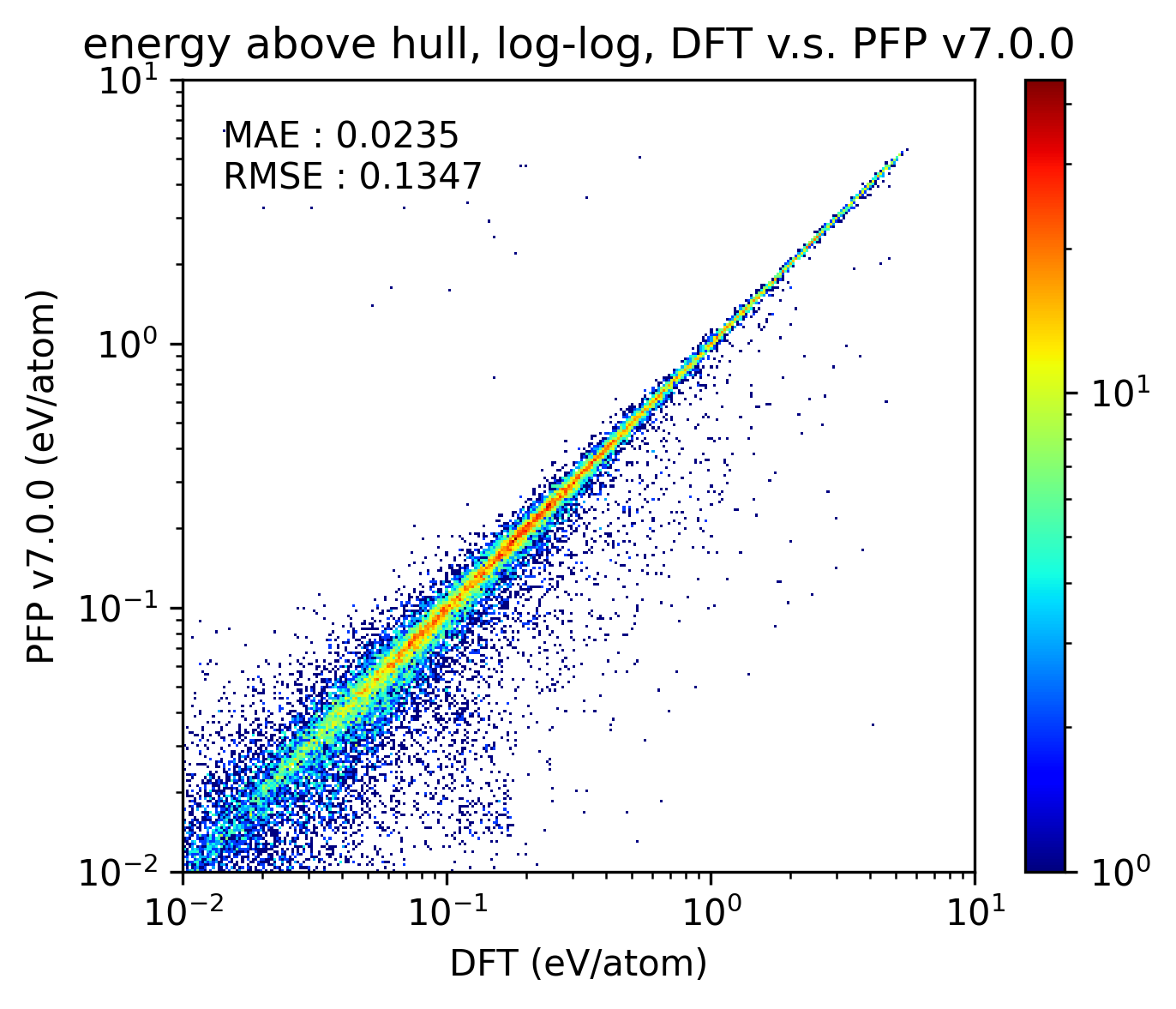 |
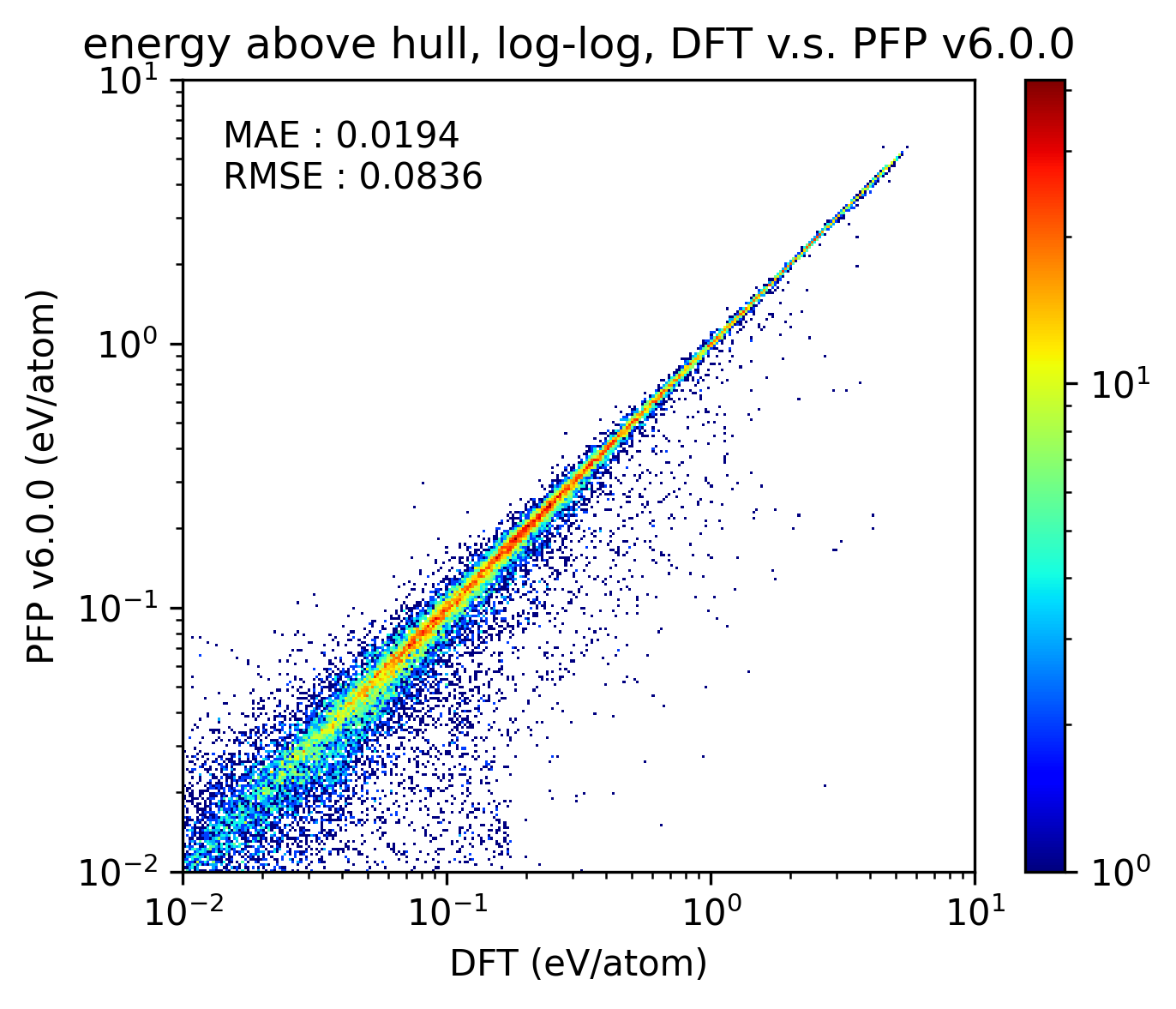 |
PFPとDFTとでのenergy above hull (Ehull)の差をプロットしたものが以下の図になります。
それぞれ、DFTでのenergy above hullが 1 eV/atom以下、0.01 eV/atom以下の構造のみに絞ってプロットしています。
両者ともv7.0.0ではv6.0.0と比較してMAEがわずかに悪化しています。
また、0.01 eV/atom以下の構造に絞った場合の方がMAEが小さく、PFPではより安定な構造はより高い精度でenergy above hullを計算することができることがわかります。
| Ehull ≤ 1 eV/atom | Ehull ≤ 0.01 eV/atom |
|---|---|
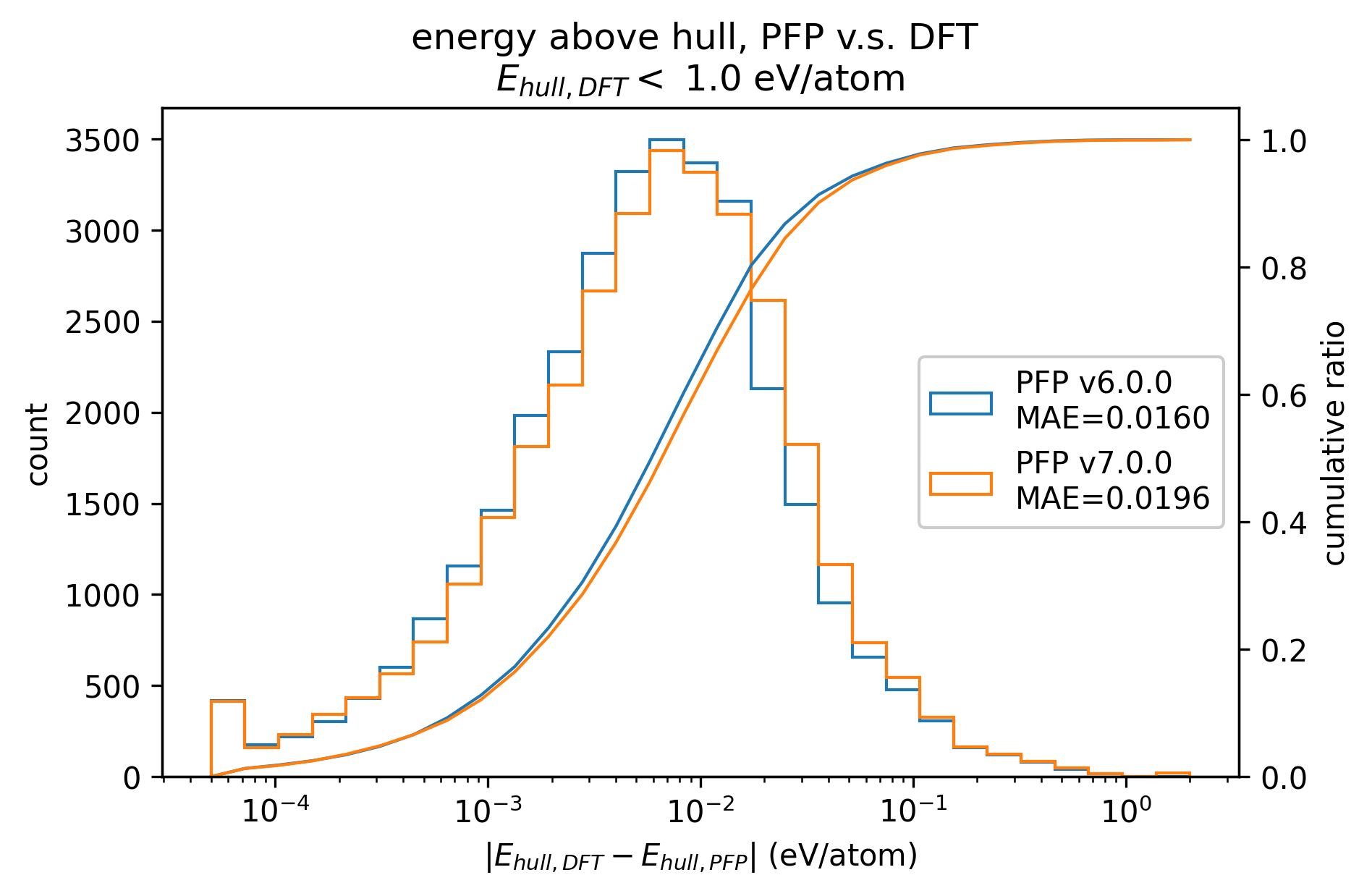 |
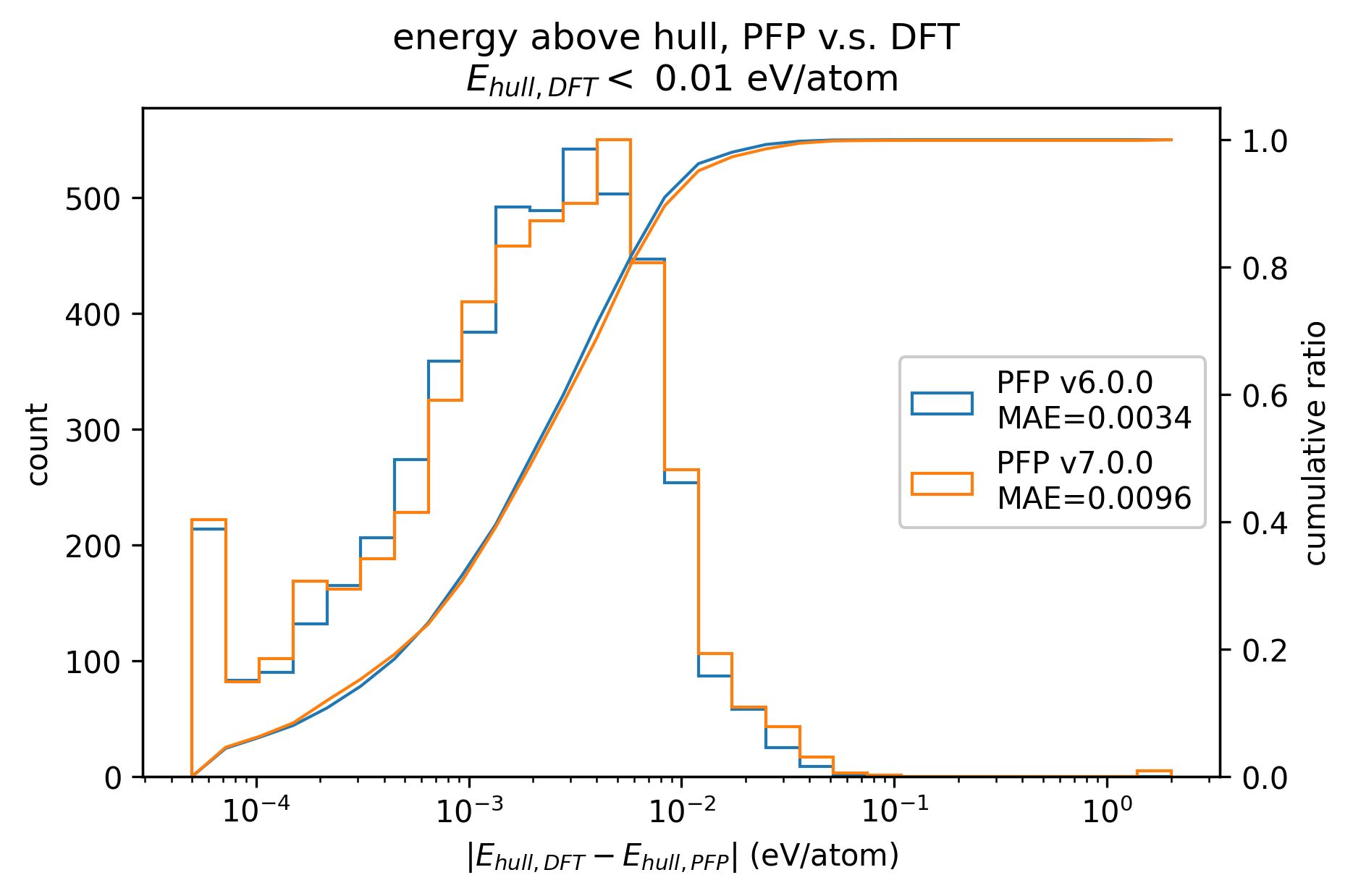 |
高密度構造における安定性検証
実際よりも高密度な構造に対してポテンシャルエネルギー曲面(Potential Energy Surface, PES)が不安定であることが、v5.0.0やそれ以前のversionのPFPの課題でした。
このような構造は通常は存在しませんが、超高圧下でのシミュレーションや、結晶構造探索などにおける機械的な構造生成で出現することがあります。
高密度構造でのPESの不安定により、これらの構造に対する構造最適化が収束しない、あるいは、本来の局所安定構造から大きく離れた構造に収束してしまう、といった問題が生じます。
過度に高密度な構造に対して非常に低いエネルギーが返されると、たとえばより安定でエネルギーが低い構造を探索するというタスクである、結晶構造探索を破綻させてしまいます。
COD構造
Crystallography Open Database (COD) 1に含まれる、PFP v3.0.0以降でサポートする72元素のみで構成される結晶構造を対象としました。
これらの構造をDFTを用いて構造最適化を行った後、各構造の格子長を変えて、等方圧縮におけるPESの安定を検証しました。
構造とDFTによる構造最適化の詳細は、後述のベンチマークデータセットと構造最適化の節を参照してください。
PFPでは、構造最適化に使用されたDFT条件に相当するCRYSTAL_U0を指定しました。
0.985=0.9039倍に圧縮した構造から開始し、ステップごとに格子長を0.98倍にすることを繰り返しました。
エネルギーが前のステップと比較して低下またはnanとなった場合、PESが不安定になったとみなしました。
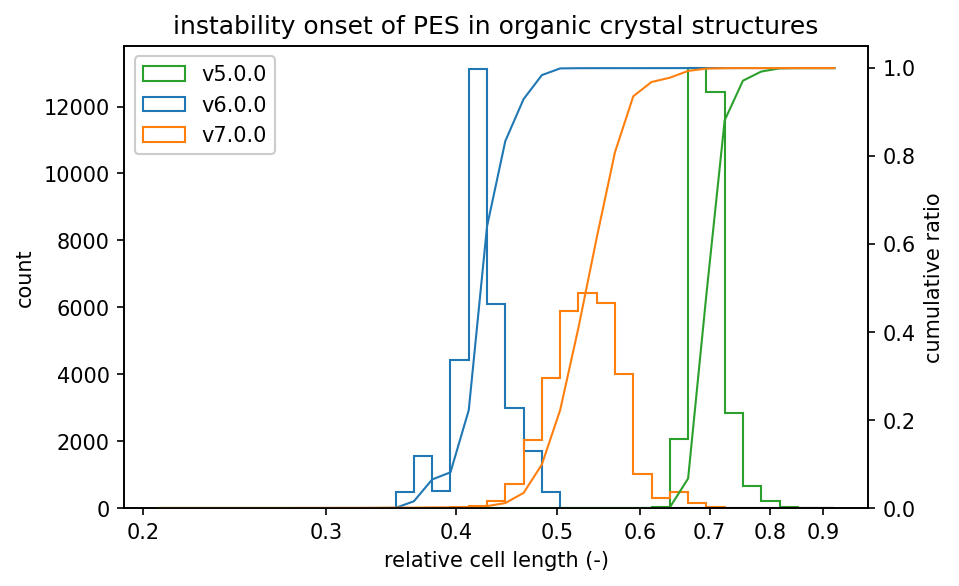
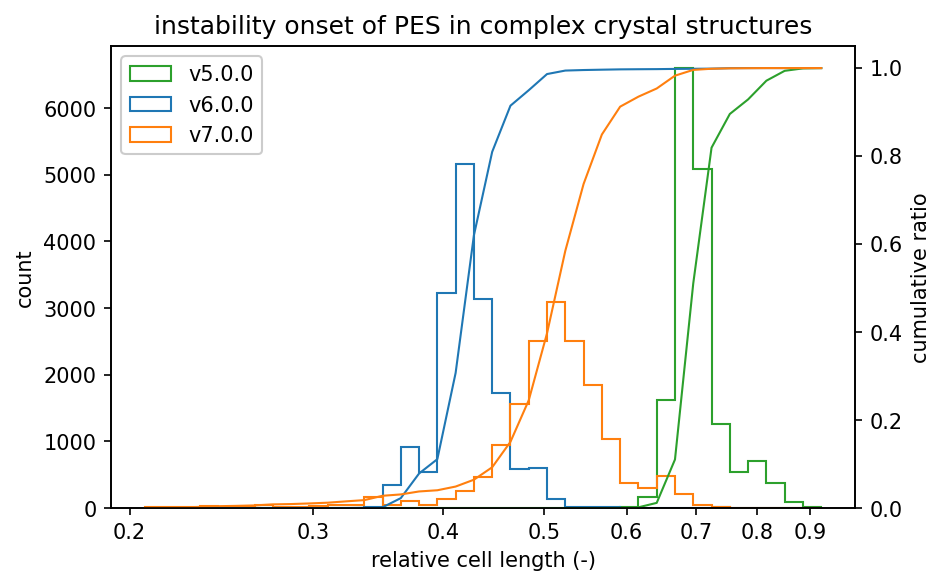
ヒストグラムは、不安定となったステップの一つ前での相対格子長の分布を示しています。
| 有機結晶 | 錯体結晶 | 無機結晶 |
|---|---|---|
 |
 |
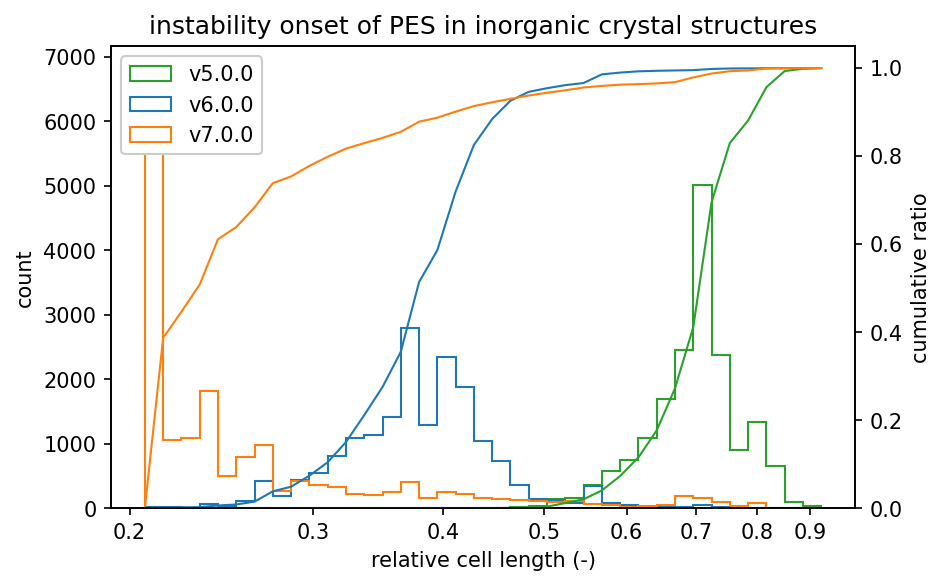 |
グラフの左側にヒストグラムが集中しているほど、高密度でのPESの安定性が高いと言えます。
有機結晶、錯体結晶、無機結晶の分類はCOD構造によるベンチマークと同様に行っています。
有機結晶と錯体結晶については、v7.0.0の高密度でのPESの安定性はv6.0.0よりは幾分低いもののv5.0.0の結果よりは向上しています。
また無機結晶についてはv7.0.0の性能がv6.0.0よりもさらに改善しています。
有機結晶の場合、v5.0.0では格子長を0.77倍まで圧縮すると2%の構造に対してPESが不安定になり始ます。
一方で、v7.0.0では2%の構造に対してPESが不安定になり始めるのは、格子長を0.64倍まで圧縮したときです。
無機結晶にはより多様な元素が含まれるため、ヒストグラムがより広がりを持っています。
分布が広いので有機結晶よりは不安定になるのが早いものの、v7.0.0で2%の構造に対してPESが不安定になるのは、格子長を0.69倍まで圧縮したときであり、v5.0.0での0.83倍より大きく改善しています。
参考文献
-
“Crystallography Open Database” https://www.crystallography.net/cod/ ↩↩
-
“Materials Project” https://materialsproject.org/ ↩
-
So Takamoto, Chikashi Shinagawa, Daisuke Motoki, Kosuke Nakago, Wenwen Li, Iori Kurata, Taku Watanabe, Yoshihiro Yayama, Hiroki Iriguchi, Yusuke Asano, Tasuku Onodera, Takafumi Ishii, Takao Kudo, Hideki Ono, Ryohto Sawada, Ryuichiro Ishitani, Marc Ong, Taiki Yamaguchi, Toshiki Kataoka, Akihide Hayashi, Nontawat Charoenphakdee, and Takeshi Ibuka, “Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements,” Nature Communications 13, 2991 (2022). https://doi.org/10.1038/s41467-022-30687-9 ↩
© Copyright 2024, Preferred Networks, Inc. and ENEOS Corporation
-




 URLを
URLを
コピーしました