Data-Driven Method for Discovering Low Adsorption Energy Molecules on Si Surfaces
Theme Overview
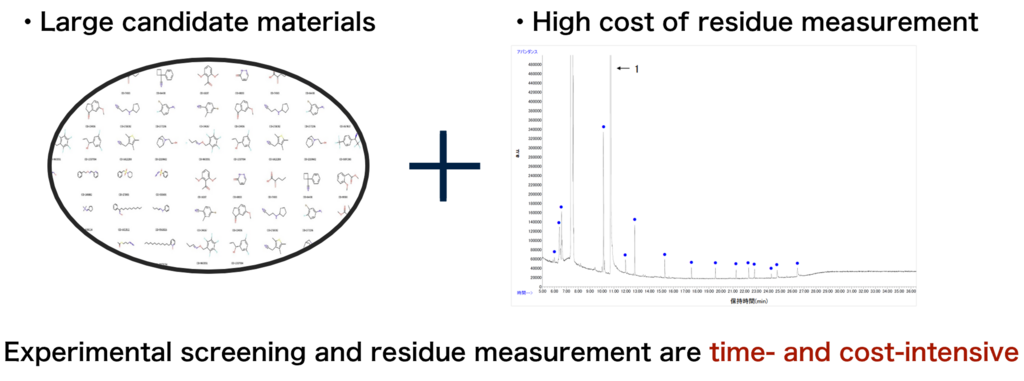
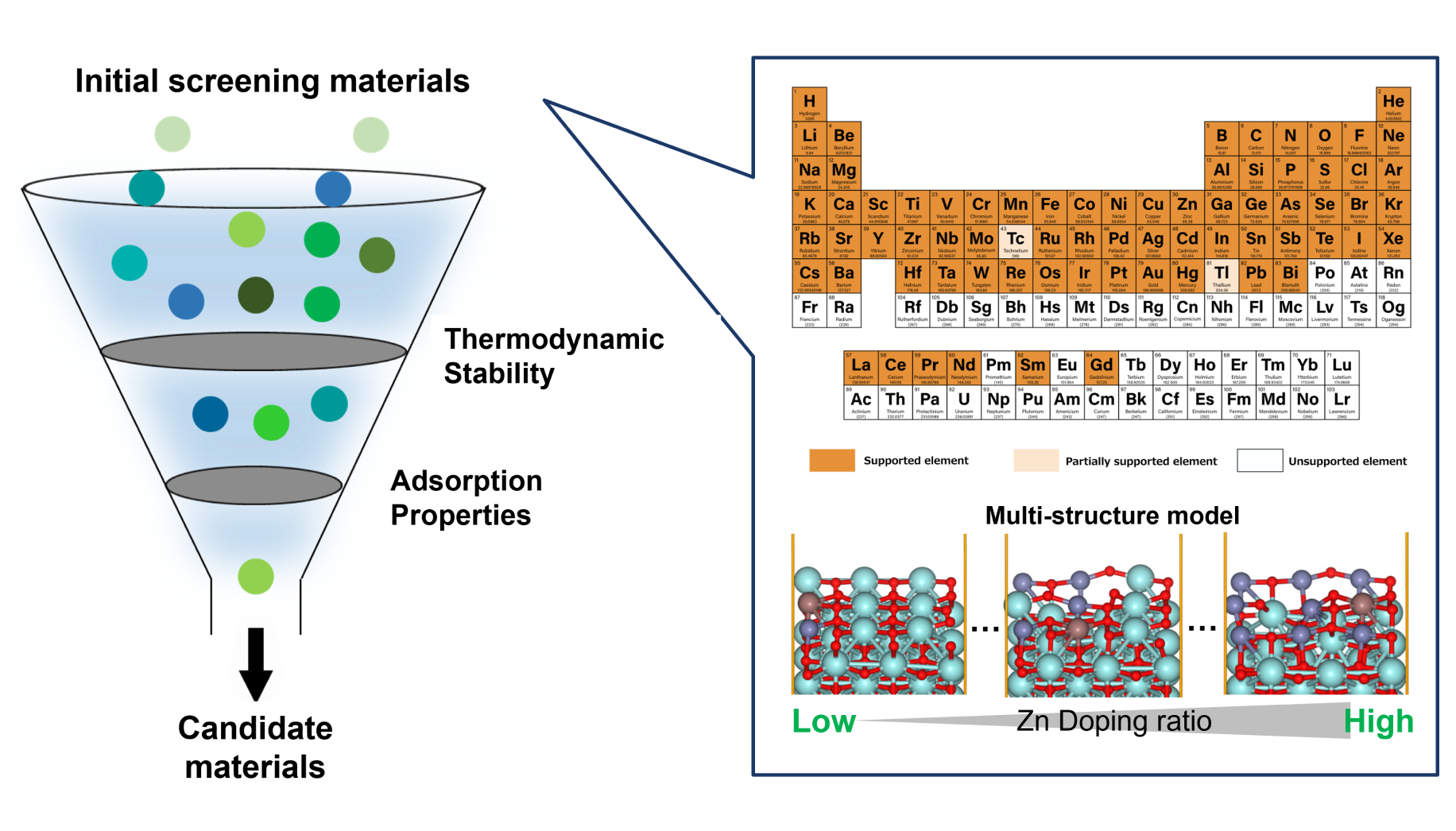
In semiconductor manufacturing processes, a high level of cleanliness is required to prevent residues from remaining on Si substrates, and the application of new chemical solutions is being considered. However, experimentally evaluating and selecting suitable low‑residue materials from an enormous number of candidates is challenging due to the substantial time and financial costs required.
Data-driven exploration frameworks that integrate sequential experimental design based on Bayesian optimization (BO) with simulation-based property calculations have been proposed as effective approaches for efficient exploration of large materials spaces [1].
In this study, a data-driven materials exploration workflow is implemented to efficiently screen candidate materials by evaluating the amount of residue remaining on Si substrates after HF treatment using adsorption energies. Within this workflow, adsorption energies are computed using Matlantis, and the resulting data are iteratively fed back to update a machine learning model, enabling efficient identification of low-residue candidate materials from an extensive candidate space.
Calculation Models and Methods
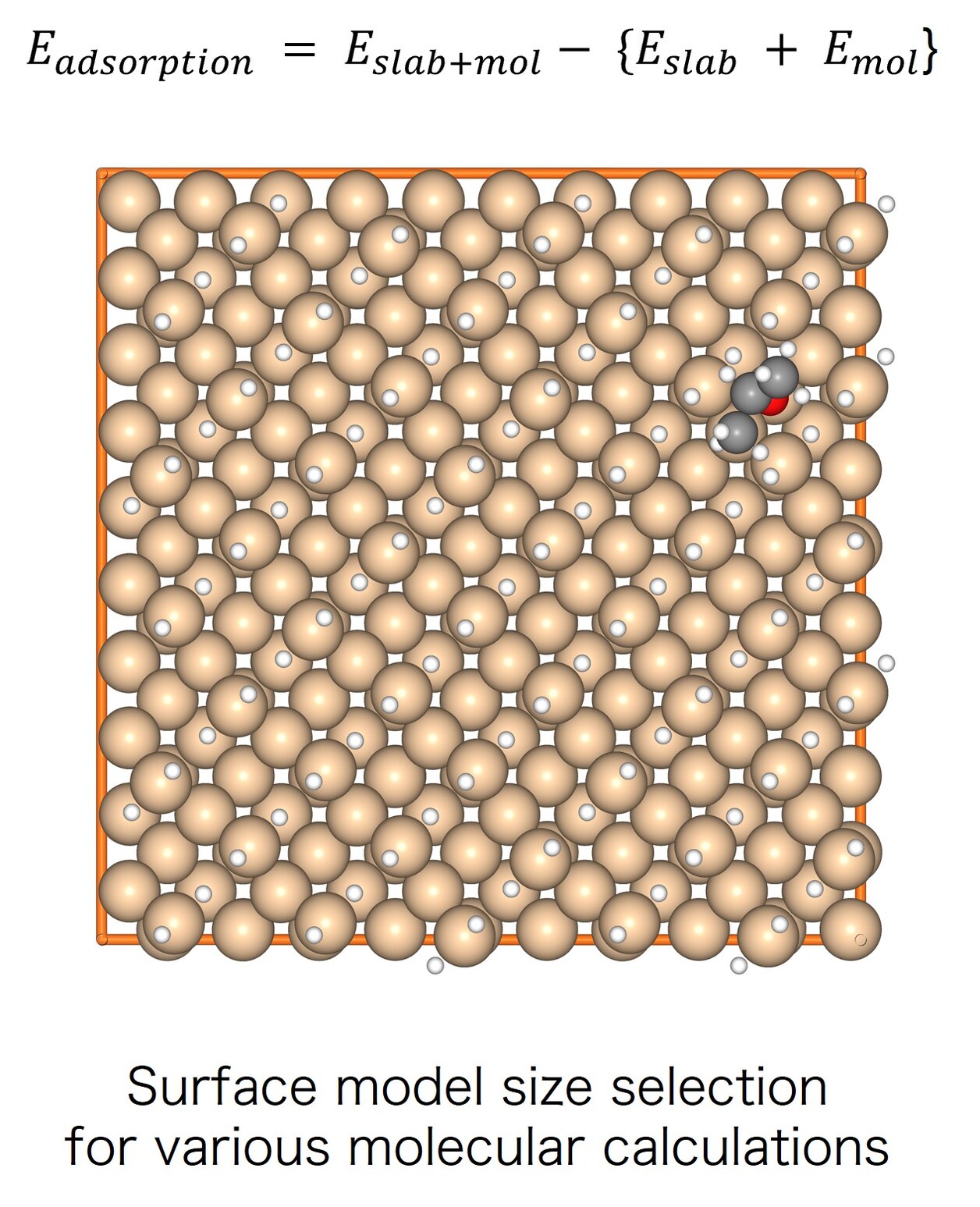
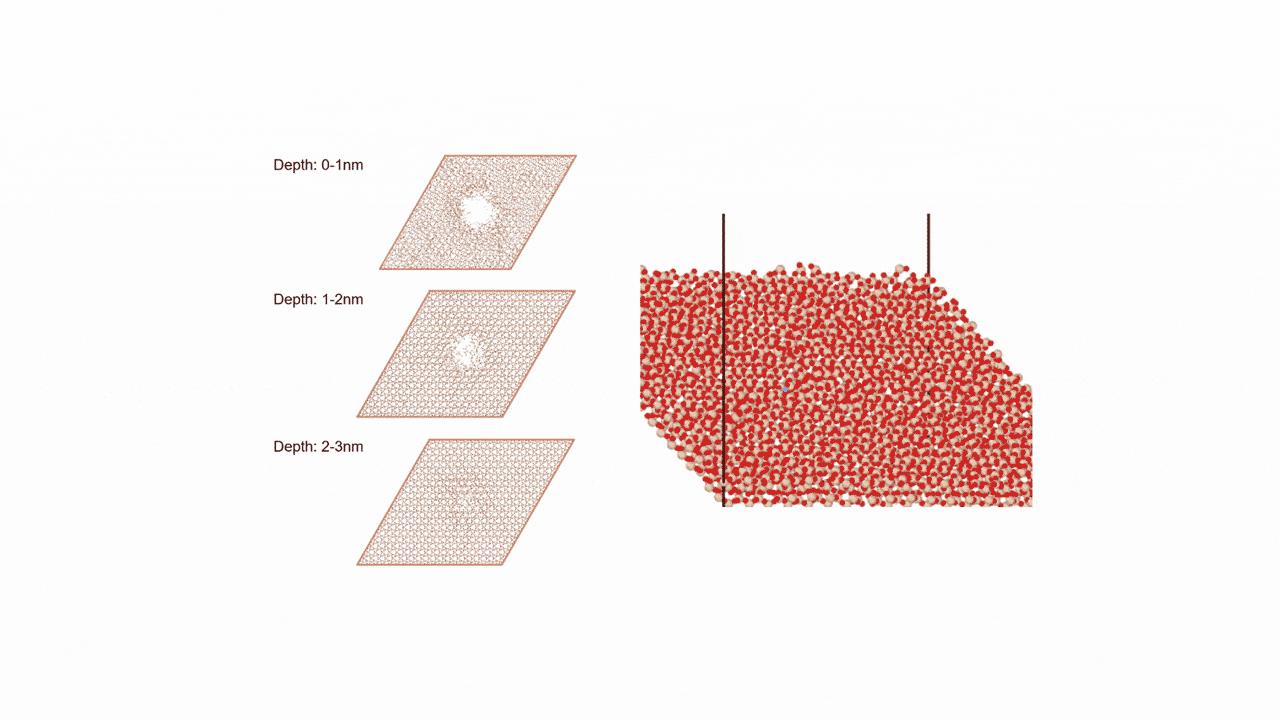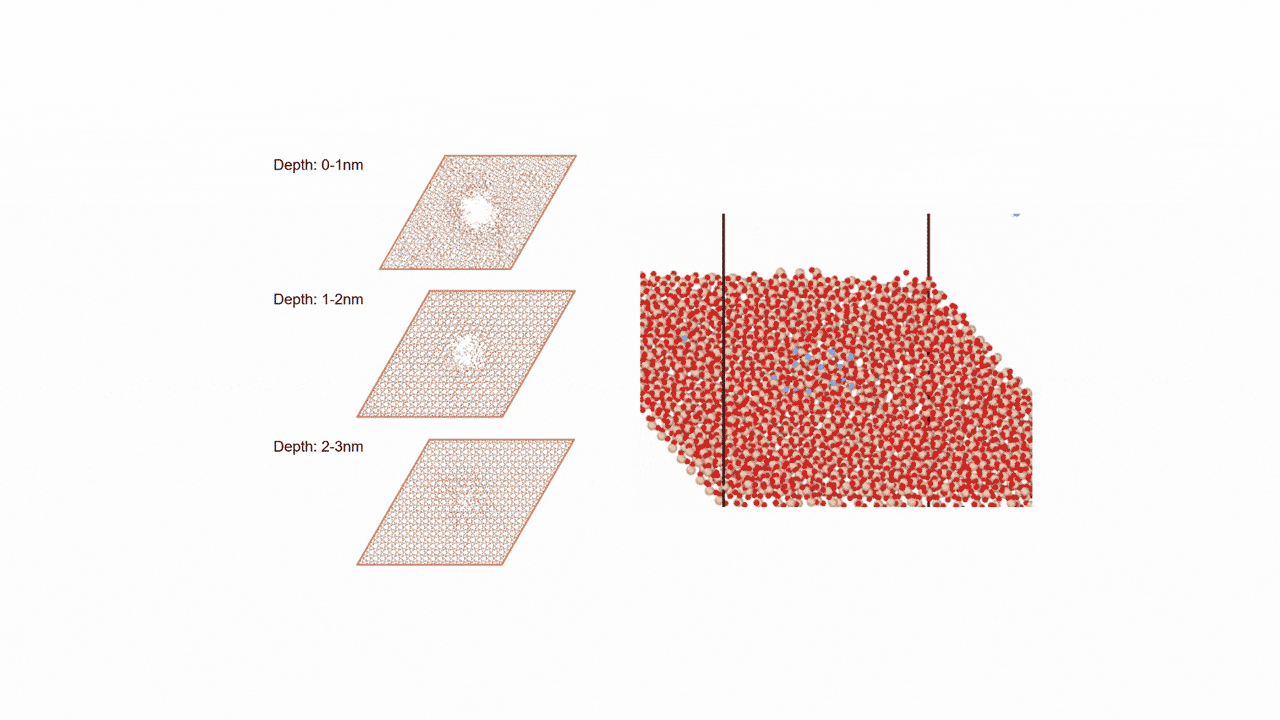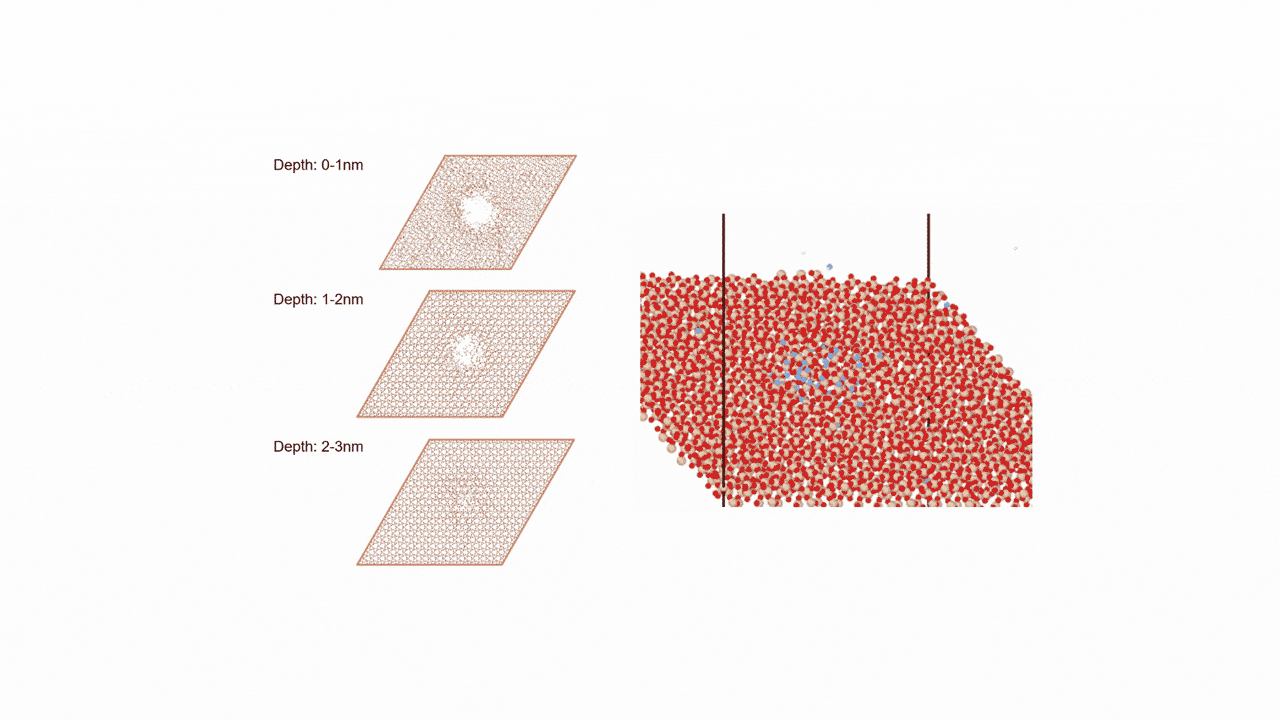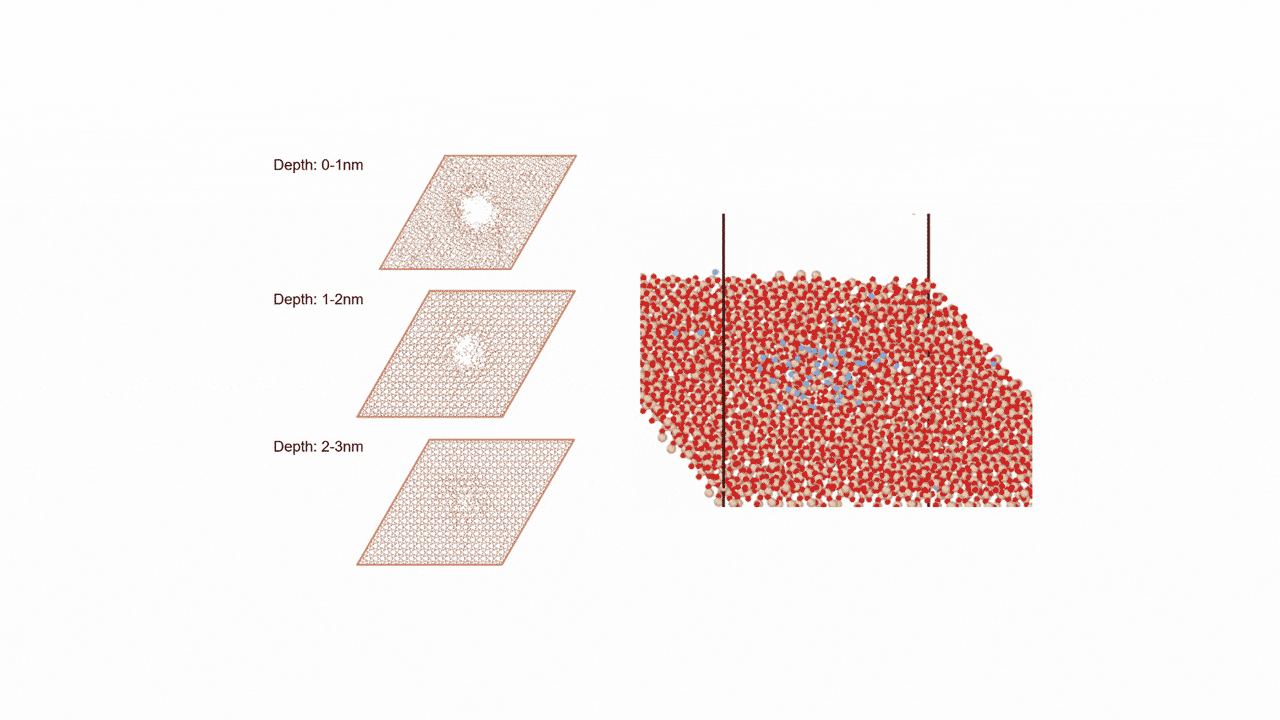
Molecules exhibiting lower adsorption energies on Si surfaces were assumed to correspond to materials that leave fewer residues on Si substrates. A hydrogen-terminated Si surface model, representing the post-HF treatment condition, was constructed using the Atomic Simulation Environment (ASE), and adsorption energy calculations were subsequently performed.
Given the structural diversity of the candidate materials, adsorption structure searches were conducted using Bayesian optimization implemented in Optuna to identify the most stable adsorption configurations, from which the corresponding adsorption energies were obtained [2].
The adsorption energies were calculated following the slab + molecule energy scheme described in the Atomistic Simulation Tutorial, using the following equation [3]:
Results and Discussion
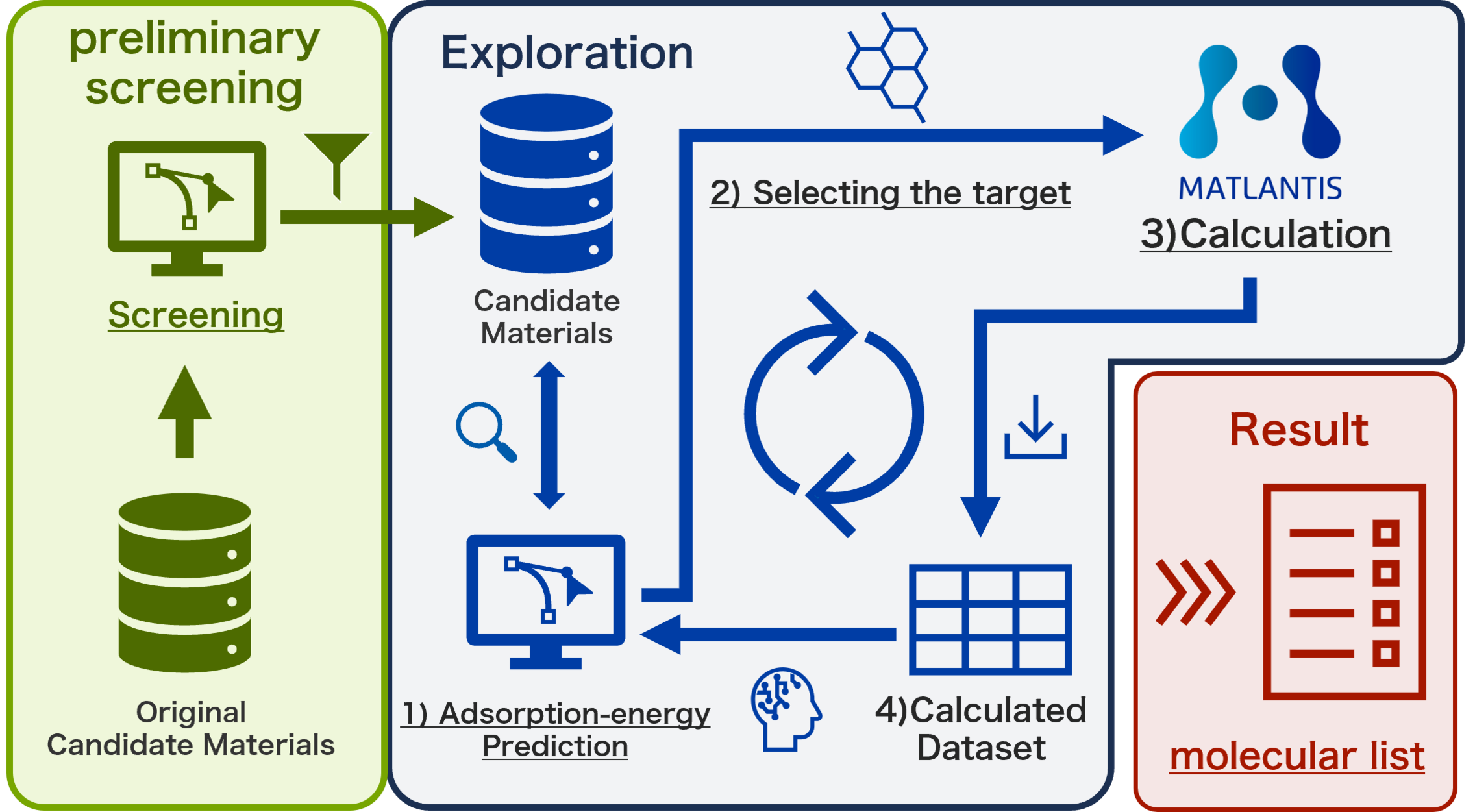
Figure 3 illustrates an overview of the data-driven exploration workflow, in which adsorption energy calculations are automated using Matlantis and sampling is performed using a machine learning model. For each material, molecular representations were generated using RDKit descriptors [4] and Force-Field Kernel Mean descriptors [5], which were subsequently used to construct an adsorption energy prediction model.
Prior to initiating the automated exploration, an initial adsorption energy dataset was generated using Matlantis, and candidate materials were extracted from the original material pool through preliminary screening.
The workflow performs prediction and selection in the following sequence:
1. Adsorption energy prediction model ;
The initial dataset, candidate materials, and model type are first specified, after which an adsorption energy prediction model is constructed using the initial dataset.
2. Selection of the next molecule for calculation ;
Predictions are performed using the constructed adsorption energy prediction model, and a single molecule is selected from the candidate materials for the next calculation based on the exploration algorithm.
3. Adsorption energy calculation ;
The most stable adsorption structure of the selected molecule is identified using Matlantis via the automated execution program, and the corresponding adsorption energy is calculated.
4. Update of the calculated dataset ;
The computed adsorption energy is added to the dataset, and the adsorption energy prediction model is retrained using the updated dataset.
(Return to Step 1)
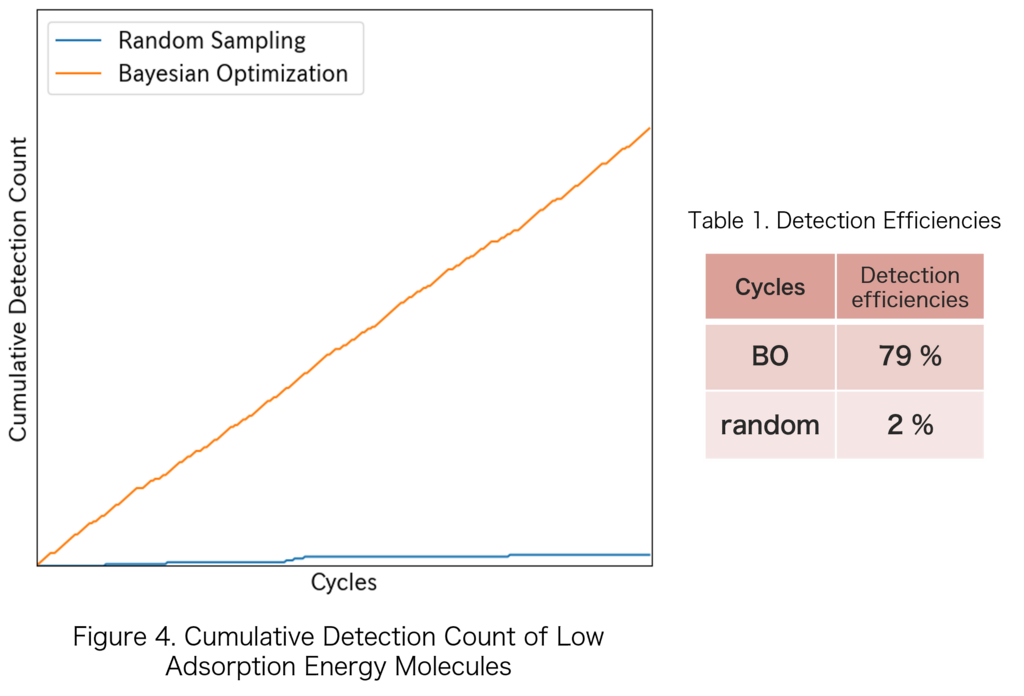
Figure 4 shows the cumulative number of detected low-adsorption-energy molecules as a function of the cycle number. A steeper slope indicates higher detection efficiency, reflecting more effective identification of candidate materials with low adsorption energies. The corresponding detection efficiencies are summarized in Table 1.
In this study, the detection efficiencies of two sampling strategies, Bayesian optimization (BO) and random sampling, were compared. In the random sampling approach, one molecule was randomly selected from the original candidate materials at each cycle.
BO enabled the identification of candidate materials exceeding the internal threshold from the early stages of the cycles and demonstrated a 39-fold improvement in screening efficiency compared with random sampling. This improvement is considered to arise from the combined effect of the preliminary screening and the selection based on the exploration algorithm.
The application of the present data-driven exploration workflow for identifying low-adsorption-energy molecules on Si surfaces is expected to facilitate the discovery of candidate materials beyond those previously selected by experimental approaches, thereby enabling the identification of new materials.
Computational Details
| Items | Details |
| Number of slab atoms | 500 atoms ~ |
| Kind of atoms | H , B , O , C , N , F , Si ,P ,S etc... |
| PFP | Ver 5.0.0 , PBE+D3 mode |
References etc.
Reference
[1] Nanjo, S., et al., npj Comput Mater 11, 16 (2025).
[2] Takuya, Akiba., et al., Optuna: A Next-generation Hyperparameter Optimization Framework. 2019
[3] Slab + mol energy — Atomistic Simulation Tutorial
[4] RDKit: Open-source cheminformatics. https://www.rdkit.org
[5] Kusaba, M., et al.,Phys. Rev. B 108: 134107 (2023)
Related Documents
Hiroe Murashima et al., The 72nd Spring Meeting of the Japan Society of Applied Physics, 15p-P06-5
Profile of the case study provider
SCREEN Holdings Co., Ltd.

Publication date of this case study: 2026.05.15
-




 URL
URL
Copied
what's new
High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
Pharmaceuticals
Calculation and Verification of Methanol Synthesis Catalysts
Methanol synthesis catalystsLarge-scale screening
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis

SiO₂ Dry Etching Simulation Using LightPFP
Semiconductor