Calculation and Verification of Methanol Synthesis Catalysts
Theme Overview
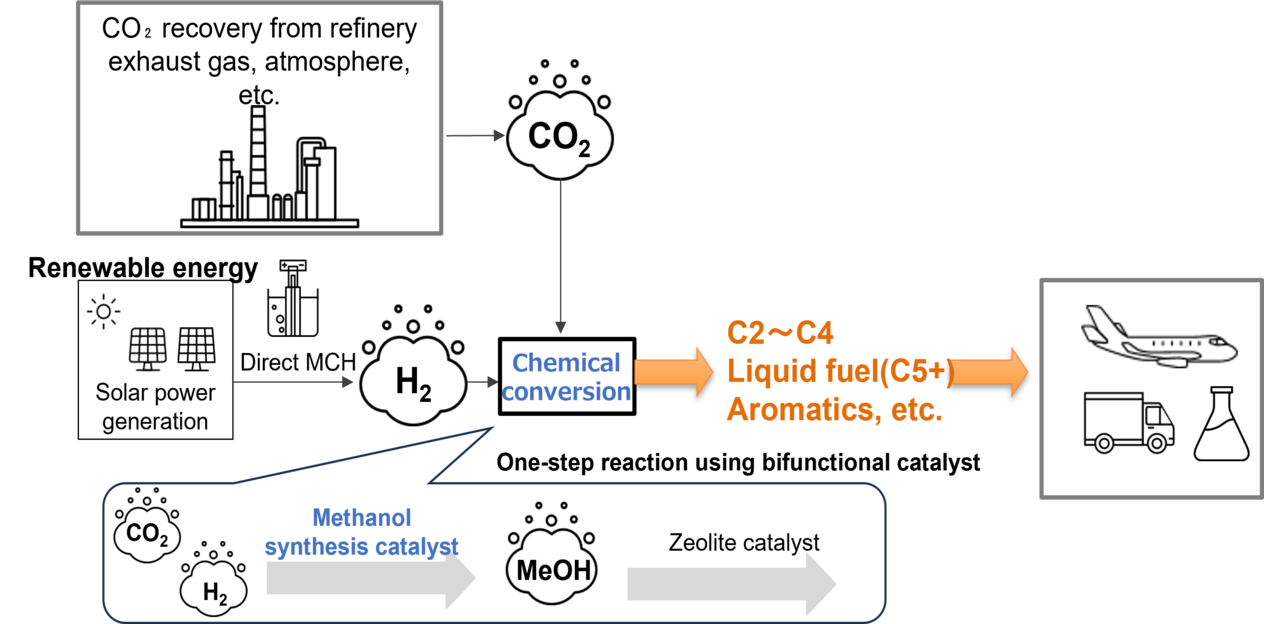
The realization of a carbon-neutral society requires essential technologies for producing chemicals and fuels using CO2 as a feedstock. Among these, methanol synthesis via CO2 hydrogenation is considered a key technology for producing major chemical feedstocks from renewable energy-derived hydrogen and CO2.
For practical applications, bifunctional catalysts that combine methanol synthesis catalysts with zeolite catalysts to enable one-step conversion into valuable chemicals have attracted significant attention. However, a critical challenge arises from the mismatch in optimal operating conditions: while zeolite catalysts require high temperatures above 300 °C for activation, conventional methanol synthesis catalysts suffer from increased side reactions at high temperatures, particularly CO formation, which leads to a decrease in methanol yield.
In this study, we focused on ZnZrOx catalysts[1] and conducted a systematic exploration of promising dopant candidates that can suppress side reactions (CO formation) even at high temperatures while maintaining high methanol selectivity, thereby achieving higher methanol yields. Specifically, we developed original multi-structure models capable of accurately reproducing complex surface structures and performed screening based on these models to identify optimal dopant candidates for ZnZrOx catalysts. Experimental validation demonstrated that the identified catalysts significantly improved methanol synthesis yields compared to undoped ZnZrOx particularly in high-temperature ranges.
Calculation Models and Methods
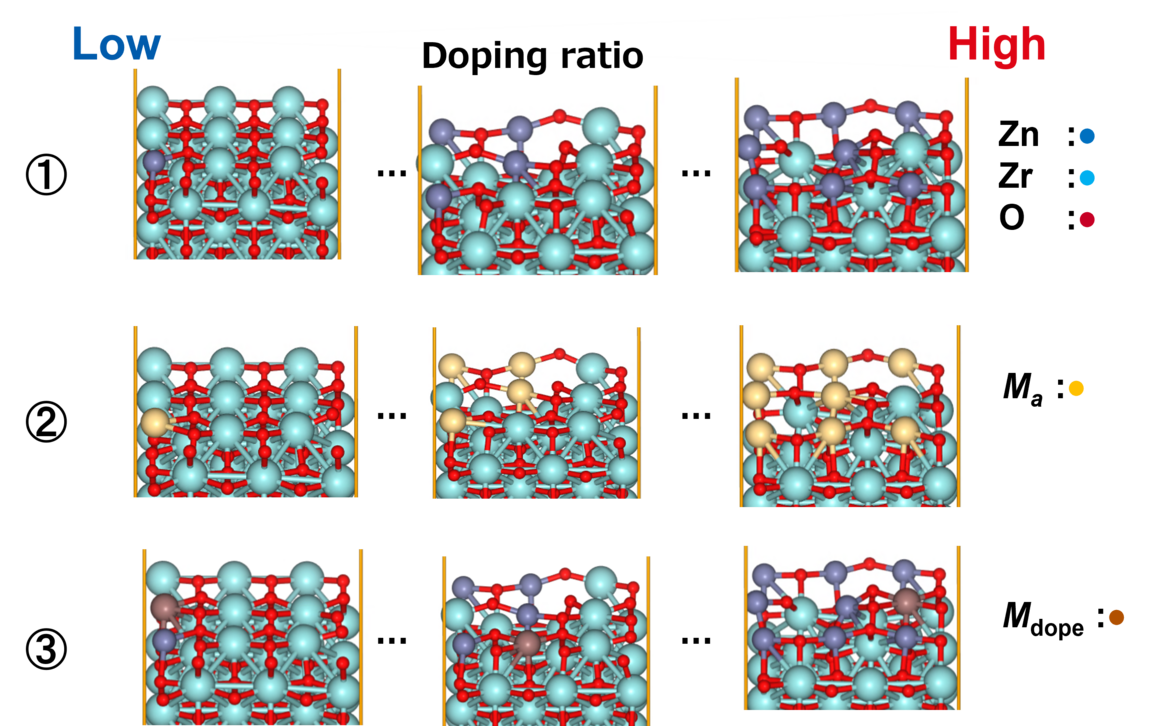
Construction of Multi-Structure Models
ZnZrOx catalysts possess complex surface structures, and a single structure model does not necessarily enable accurate prediction of experimental behavior.
To comprehensively account for diverse surface states, we constructed three more realistic models. Each of the nine slab models shown in Fig. 2 represents a single structure configuration, and these models were combined to build the overall model. Hereafter, we refer to this constructed model as the "multi-structure model."
- ①ZnZrOx(Base Model)
This model features varying Zn element ratios on the surface. We verified whether it could correctly represent the IR spectra of reaction intermediate molecules adsorbed on the catalyst. - ② Ma-ZrO x
This model is a modified version of the base model in which the Zn elements on the surface are completely replaced with other Maelements (Ga, Cd, In, etc.). We verified whether the adsorption energy could be reproduced as an indicator for evaluating catalytic activity. - ③ Mdope-ZnZrO x
Different elements in the base model Mdope This model is doped with trace amounts of (Pd, Pt, Cu, etc.). It is more in line with actual catalyst development and was used for screening (see next section for details).
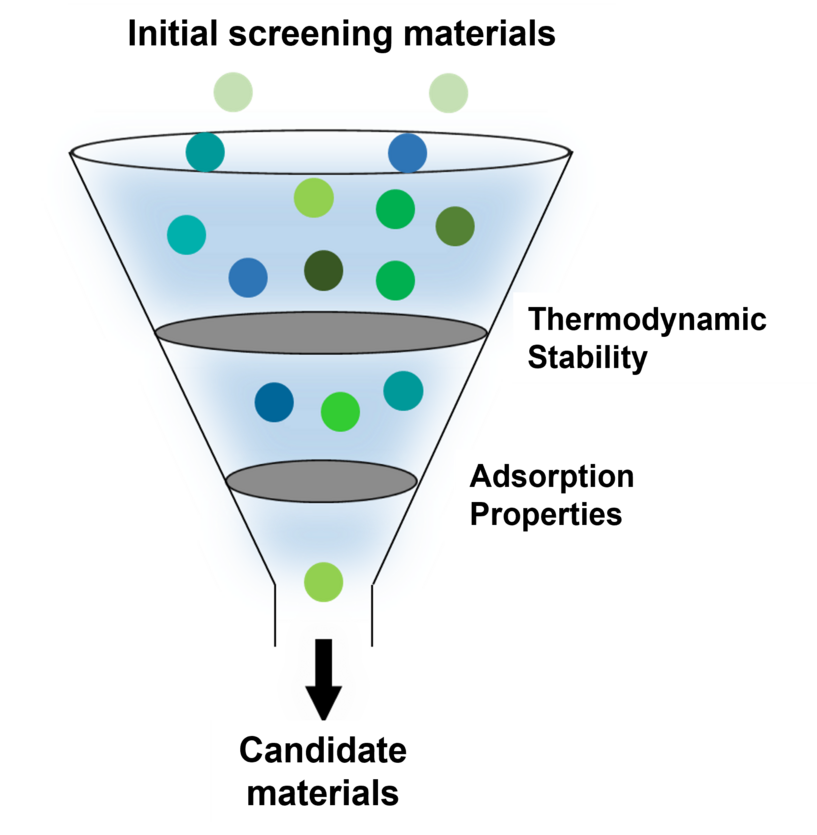
Large-Scale Screening Approach: Computation and Experimental Verification
Step 1: Validation of the multi-structure models (Models ① and ②)
We first examined whether the multi-structure models can reproduce experimental observations required for catalyst development, in comparison with single structure models.
- For Model ①, to investigate the effect of surface structures with different Zn ratios, we calculated IR spectra of reaction intermediates on the catalyst surface and compared them with experimental data.
- For Model ②, to evaluate changes in catalytic activity induced by surface metal substitution, we assessed the adsorption energies of the reactant (CO2*) and the intermediate (CO*).
Step 2: Large-scale screening (Model ③)
Using Model ③, which represents heteroatom-doped ZnZrOx with varying Zn ratios, we conducted a systematic screening over 72 elements supported in PFPv5.
- Evaluation of synthesizability (thermodynamic stability):
From a practical catalyst design perspective, we evaluated the stability of heteroatom-doped systems using bulk structures of Model ③. - Evaluation of catalytic activity (adsorption properties):
We calculated the adsorption energies of hydrogen (H*) and the intermediate (CO*) on the catalyst surface.
Step 3: Experimental validation
Based on the screening results, we synthesized catalysts doped with promising candidate elements and experimentally evaluated their methanol synthesis yields.
Results
Validation of Multi-Structure Model ①
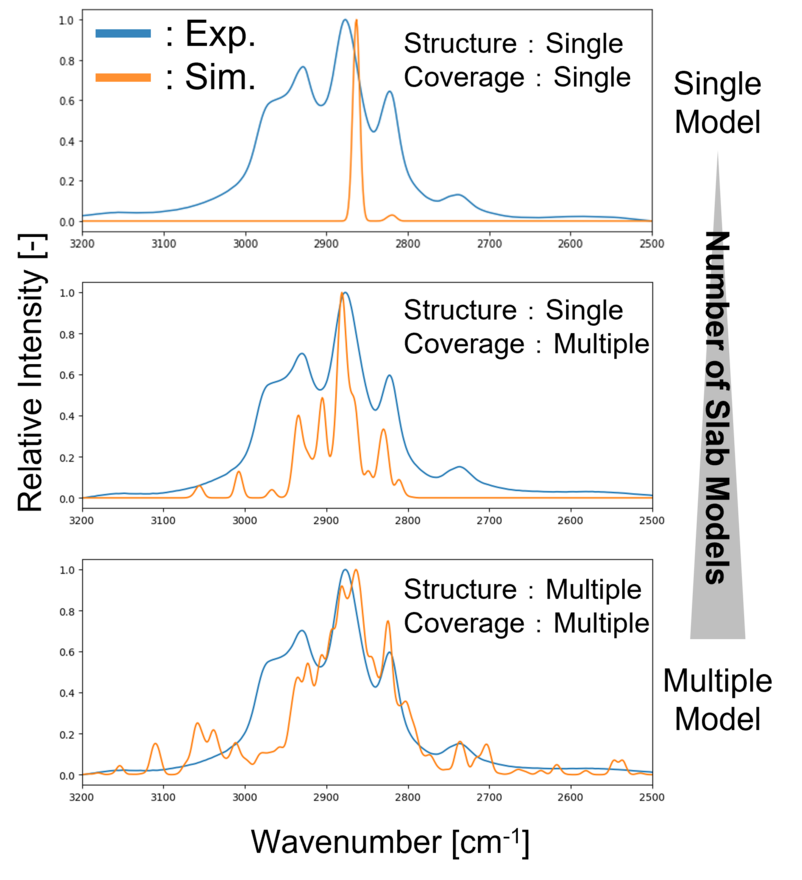
We validated whether the constructed multi-structure Model ① (ZnZrOxx base model) can accurately describe phenomena occurring on actual catalyst surfaces.
First, reaction intermediates such as CH3O* and HCOO* were adsorbed on the catalyst surface, and the IR intensities and wavenumbers of these intermediate species were calculated. The obtained results were then compared with experimental data measured by in-situ DRIFT spectroscopy (Fig. 4).
In the single-structure model with one adsorbed HCOO* molecule, the peak position of HCOO* around 2850 cm-1 was qualitatively reproduced; however, other peak positions could not be captured.
In contrast, the multi-structure model successfully reproduced the overall experimental trends by linearly combining the IR spectra of multiple structure models with different surface Zn ratios and intermediate coverages. In particular, it accurately captured the key vibrational modes of important intermediates, CH3O* (2930cm-1) and HCOO*(2876cm-1) [2].
Validation of Multi-Structure Model ②
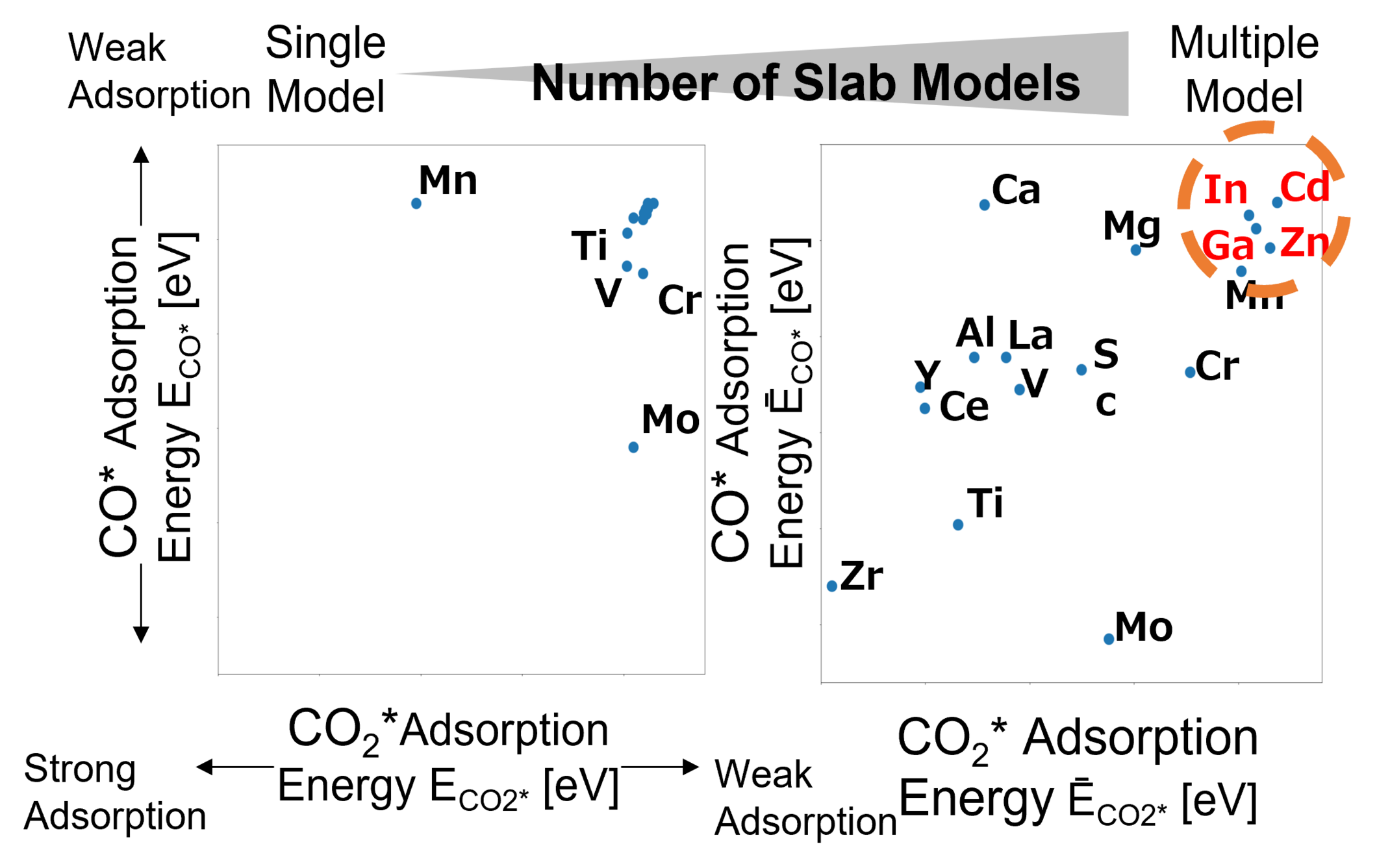
We validated whether the calculated values obtained using the constructed multi-structure Model ② (Ma-ZrOx) can serve as descriptors for predicting actual catalytic activity by comparison with experimental data reported in Ref. [3]. In the surface structure models, CO and CO₂ were placed on various adsorption sites, followed by structure optimization to identify the most stable adsorption energies, ECO* and ECO2*, within each model. In the multi-structure model, the statistically averaged values, ĒCO*、ĒCO2* weighted by dopant ratio, were used as descriptors for catalytic activity.
In the single-structure model, no clear differences in adsorption energies were observed regardless of catalytic activity, and thus the experimental trends could not be reproduced.
In contrast, the multi-structure model showed that elements reported to exhibit high activity [3] (Zn, Cd, In, and Ga) cluster within a specific adsorption energy region. This indicates that the model enables prediction of highly active catalyst candidates based on adsorption energies.
From the validation results of multi-structure Models ① and ②, it was confirmed that correlations between structure and activity on complex, realistic catalyst surfaces -difficult to capture with single-structure models- can be described with high accuracy using the multi-structure approach.
Evaluation of Thermodynamic Stability (Synthesizability)
We conducted a systematic screening of a large number of catalyst candidates with trace metal doping from the perspectives of thermodynamic stability (synthesizability) and adsorption properties (catalytic activity).
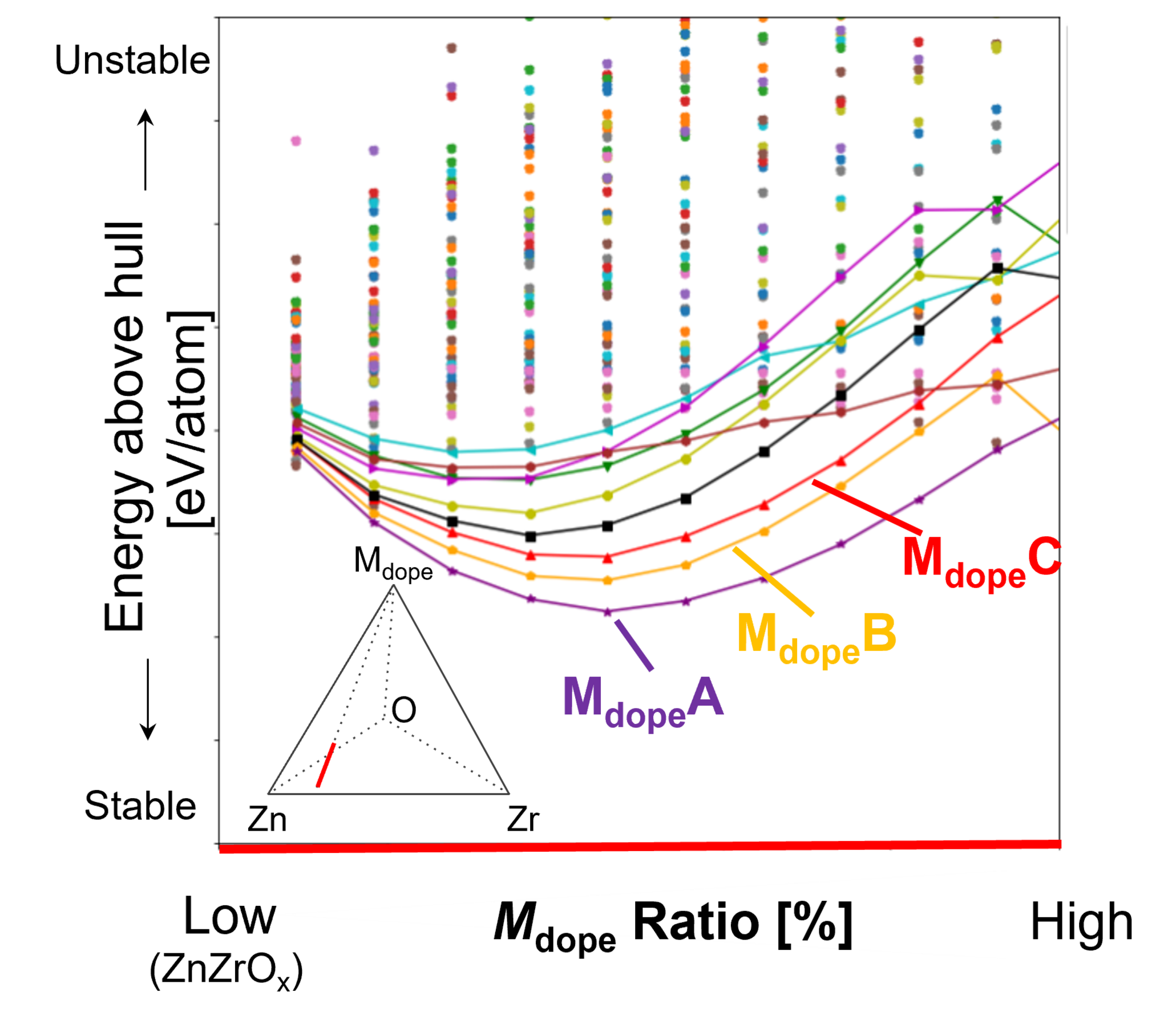
First, convex hull calculations were performed for heteroatom (Mdope)-doped ZnZrOx bulk structures. The stability (synthesizability) was evaluated as a function of dopant ratio (Fig. 6).
The results showed that, for many elements, the addition of Mdope destabilizes the ZnZrOx bulk material. In contrast, certain elements (e.g., A, B, and C) were found to stabilize the ZnZrOx catalyst when introduced in small amounts.
For these candidate elements (A, B, C, etc.), the energy above hull was improved at specific dopant ratios, suggesting the formation of more thermodynamically stable phases. Based on these findings, we proceeded to the next stage, where the surface adsorption properties were evaluated in detail.
Large-Scale Screening Using Multi-Structure Model ③
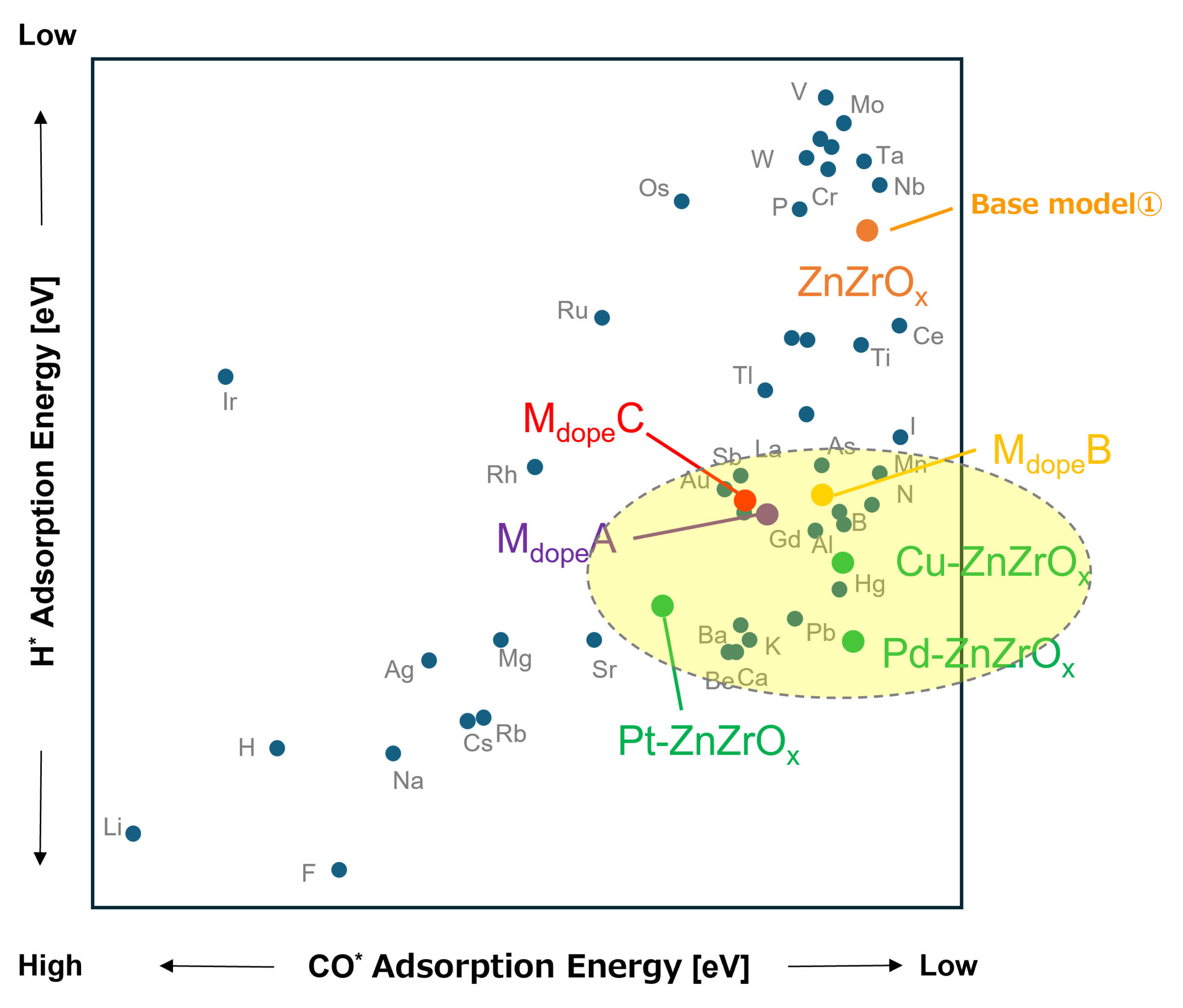
Next, using the multi-structure Model ③ (Mdope-ZnZrOx), we evaluated the averaged adsorption energies, Ē of H* and CO* (Fig. 7).
The performance was assessed relative to the base Model ① (ZnZrOx) from the following perspectives:
・Promotion of H* adsorption (contributing to enhanced CO2 hydrogenation activity)
・Minimal change in CO* adsorption (to minimize the impact on CO byproduct formation)
Among the elements screened based on the energy above hull results, ZnZrOx catalysts doped with small amounts of selected elements (A, B, and C) were found to fall within a similar region to highly active metals such as Pd, Pt, and Cu reported in previous studies. These elements were thus identified as promising candidates that satisfy the desired properties described above.
As a result of the overall screening, rare-earth elements such as MdopeA, MdopeB, Mdope were identified as the most promising dopant candidates, achieving a balance between catalyst stability and potential activity.
Results and Discussion
Based on the predictions of the multi-structure model, we synthesized the promising candidate materials identified through screening and evaluated their methanol synthesis yields.
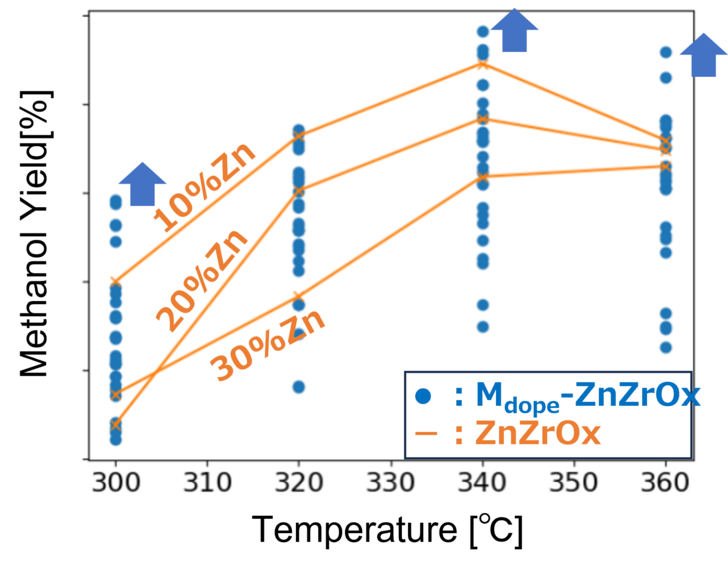
The blue data points in Fig. 8 represent the methanol yields of the newly developed catalyst candidates with varying ratios of Zn and dopants (A, B, and C) in the high-temperature range.
The experimental results are consistent with the predictions of the multi-structure model: the newly developed catalysts exhibit significantly improved methanol yields compared to the conventional ZnZrOx catalyst, particularly in the high-temperature range required for bifunctional catalysts.
・At 340 °C: up to +8% improvement in yield
・At 360 °C: up to +28% improvement in yield
The enhanced methanol yield is accompanied by suppression of CO formation, which is a key challenge at high temperatures, indicating that high selectivity is maintained.
Computational Details
Large-Scale Screening Calculation method
| Items | Details |
| Number of slab atoms | ~160 atoms |
| PFP | V5.0.0 CRYSTAL_U0 |
| Adsorption structure search | Minima hopping LBFGS Fmax[eV/Å]: 0.01 |
| Large-Scale screening calculation time※ | ~2 weeks |
References
[1] Jijie Wang, et.al., Science Advances, 3, e1701290 (2017)
[2] Kyungho Lee, et. al., Applied Catalysis B: Environmental, 304, 120994 (2022)
[3] Jijie Wang, et. al., ACS Catalysis, 9, 10253 (2019)
[4] Y. Yayama, Catalysts and Catalysis, 68(2), 93 (2026)
Profile of the case study provider
ENEOS Holdings, Inc.

Publication date of this case study: 2026.04.17
-




 URL
URL
Copied
what's new
NEW
Interfacial thermal resistance calculation at a metal–polymer interface
Interfacial thermal resistance of semiconductoradhesive materials
High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
Bio and Pharma
Data-Driven Method for Discovering Low Adsorption Energy Molecules on Si Surfaces
ScreeningMaterials InformaticsSemiconductors
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis
