This article was written by Cho, a customer success engineer at Matlantis Corporation, and was published in the February 2026 issue of the technical magazine Monthly Material Stage in a special feature titled "Improving the efficiency of materials development using AI and automated experiments." This article has also been published on our blog.

1. The History of Materials Development
Our lives have been enriched by numerous innovative materials. However, behind the development of these materials lies a daunting history of trial and error. The Haber-Bosch process, a method for synthesizing ammonia from nitrogen and hydrogen, is considered one of the greatest inventions of the 20th century. However, the catalyst essential for commercializing this technology was only discovered by Alwin Mittasch after experimenting with approximately 2,500 different materials [1].
Given this background, materials development is often perceived as being heavily experiment-driven. However, theoretical approaches have also played an important role. First-principles calculations, such as density functional theory (DFT), can calculate the energy of materials, the forces acting on each atom, and their electronic states. Using these methods, it is possible to identify stable structures of materials, analyze dynamics at the atomic level, and explore chemical reaction pathways. In this article, we refer to research approaches that use these methods as "computational chemistry." computational chemistry has the potential to significantly streamline the development process by predicting the properties of materials prior to experimentation and narrowing down promising candidates.
However, computational chemistry is not always utilized in the field of materials development. At its core lies a deep-seated "trade-off between accuracy, computation time, and universality": highly accurate methods such as DFT require extremely long computation times, while fast methods like classical potential analysis have limitations in accuracy and universality.
In recent years, machine learning (ML) has been attracting attention as a technology that can break this stalemate. ML, which has brought about breakthroughs in natural language processing and image recognition, is being applied to computational chemistry to attempt to achieve both high-precision and high-speed calculations. This is Machine learning Interatomic potential (MLIP).
This article reviews previous computational chemistry methods and details how machine learning is overcoming the trade-offs between accuracy, computation time, and universality, fundamentally transforming the paradigm of materials development. It explains the emergence of MLIP, which dramatically improves computation speed while maintaining predictive capabilities by learning from DFT calculation results, and its advanced form, Universal Machine Learning Interatomic Potential (uMLIP), which is currently emerging as a game-changer. Finally, it explores the cutting edge of materials development using uMLIP. This is not merely a matter of accelerating computational chemistry; it marks the dawn of a new era in which AI redefines the materials development process.
2. Conventional computational chemistry methods and their challenges
As discussed above, a deep-seated challenge exists in the "trade-off between accuracy, computation time, and universality" for computational chemistry to be widely used in materials development. In this chapter, we will first examine DFT and classical potential, which are representative of conventional computational chemistry methods, and then detail the problems that each of them faces.
2.1 DFT
DFT is one of the main methods that forms the basis of computational chemistry. DFT is a method that uses the electron density of a system to calculate the energy and the forces acting on each atom. Although the accuracy of the calculation depends on the exchange-correlation functional used, it is possible to obtain highly accurate results that reproduce experimental results, and it has been used to predict the electronic properties of solid materials and evaluate the stability and reactivity of molecules. Furthermore, the forces acting on atoms obtained by DFT can be used to perform various analyses. Here, we introduce the molecular dynamics (MD) method, which simulates the time evolution of a system, and the nudged elastic band (NEB) method, which explores chemical reaction paths.
- MD method: By numerically solving the classical equations of motion mechanics for each atom, time-series data such as atomic coordinates, velocity, energy, etc. can be generated. By analyzing this data, physical properties such as the diffusion coefficient and radial distribution function can be calculated.
- NEB method: A method for predicting reaction paths from one stable state to another. It can clarify the structural changes in the system during the transition process and estimate the activation energy that acts as a barrier to the reaction.
Combining DFT with these methods makes it possible to calculate a wide variety of physical properties with high precision. However, DFT calculation are time-consuming, as they increase in proportion to at least the cube of the number of electrons in the system. In particular, the MD method requires DFT calculations at each time step of the simulation, and generally only handles a few hundred atoms, with time scales limited to the picosecond range. This makes it difficult to handle many of the phenomena that are important in materials development.
2.2. Classical potential
To avoid the long computation time of DFT, "classical potentials," which describe interatomic interactions with empirical functions, have also been widely used. A variety of classical potentials have been developed depending on the material and phenomenon being studied, such as the EAM potential applicable to metallic materials, the Tersoff potential applicable to covalent materials, and the ReaxFF which can describe chemical reactions. MD methods using classical potentials are extremely fast, making it possible to track simulations on the scale of millions of atoms in microseconds. However, this convenience comes with two major limitations. First, there is the issue of accuracy. Because the interactions are approximated by simple functional forms, the prediction accuracy is inferior to that of DFT. Second, there is a lack of universality. Many potentials have parameters tuned for specific material systems and cannot be applied to different material systems. Since the potential function needs to be readjusted every time the simulation target changes, it is difficult to apply it to comprehensive material screening.
Thus, methods using DFT have hindered high-throughput materials development due to their long computation time, while methods using classical potentials have been hindered by their low accuracy and universality. Therefore, in most cases to date, the opportunities for computational chemistry to contribute to materials development have been limited, and experimental verification has often preceded the practical materials development.
3. Machine Learning Interatomic Potentials (MLIP)
3.1 Basic Concepts of MLIP and Historical Breakthroughs
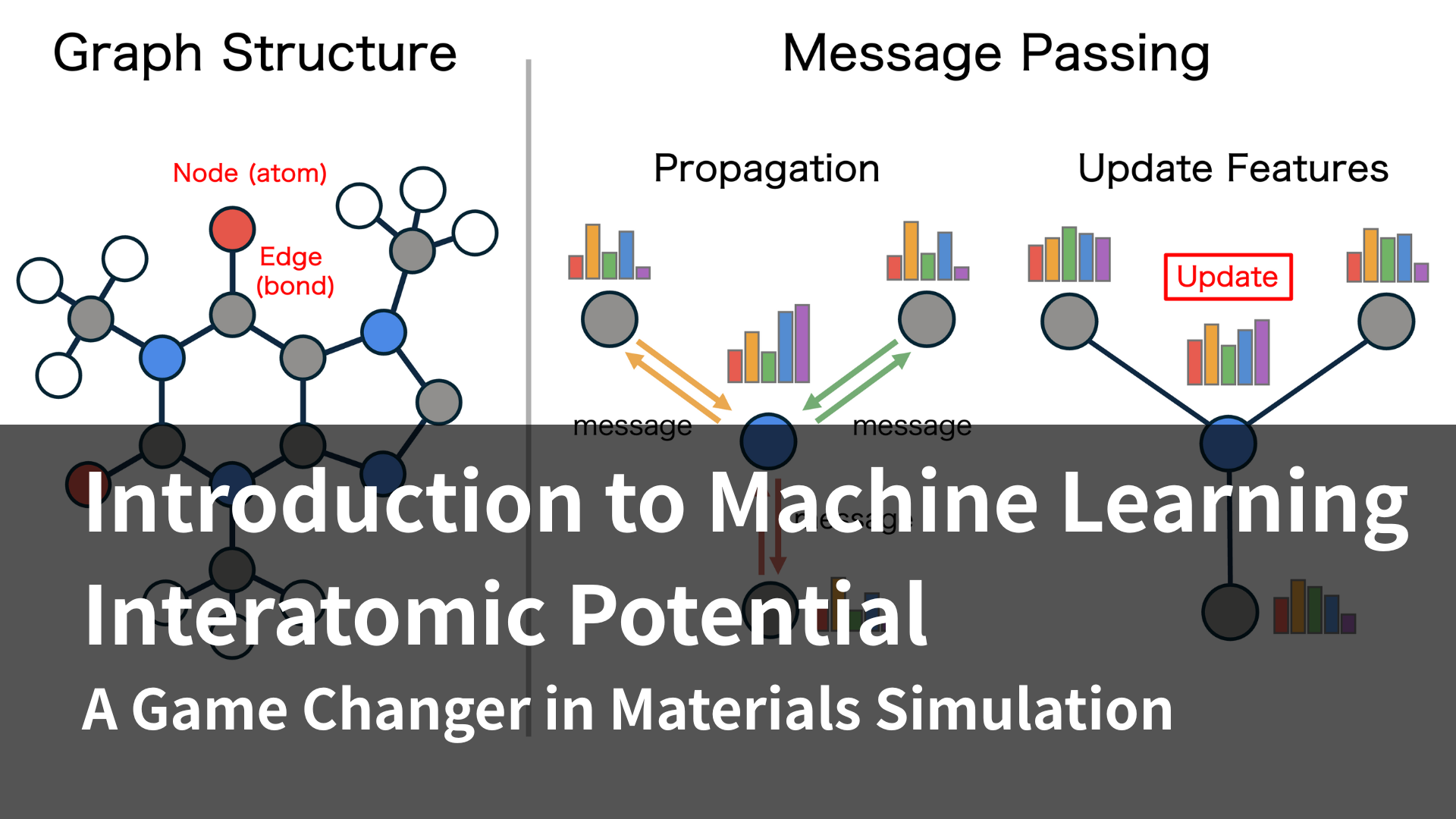
MLIP emerged as an approach to fundamentally resolve the trade-off between accuracy and efficiency that traditional computational chemistry faced. The basic principle of MLIP is to prepare a large number of pairs of input (atom types and coordinates) and output (energy and force calculated by DFT calculations, etc.), and train an ML model on the relationship between them. Once trained, the model can predict energy and force much faster than DFT calculations, making it possible to perform MD and NEB methods at high speed while maintaining accuracy comparable to DFT calculations.
Neural networks (NNs) are frequently used as the architecture for MLIPs. NNs have high expressive power and are inherently well suited to differentiation. In physics, the forces acting on atoms are calculated by differentiating energy with respect to atomic coordinates. NNs are well suited to this calculation because they can perform this calculation efficiently. Research on MLIP began as early as 1995 [2], but early models had several limitations: (1) they could only handle a fixed number of atoms, and (2) they could not guarantee physical symmetries, such as translational and rotational symmetry, for any system. While attempts were made to improve the model by applying symmetry-accepting transformations to atomic coordinates [3], a breakthrough came in 2007. The MLIP architecture, proposed by Behler and Parrinello [4], first describes the local environment of each atom using symmetry functions. Next, parameter-sharing subnetworks infer the energy of each atom and sum these to calculate the overall energy of the system. This architecture, in which the energy of the entire system is the sum of the energies of each atom, makes it applicable to systems with a different number of atoms than the training data. They then showed that an MLIP model trained using the results of approximately 8,200 DFT calculations could calculate the radial distribution function of molten silicon with the same accuracy as DFT calculations, but hundreds of thousands of times faster for a 64-atom system, demonstrating that MLIP could become a powerful simulation tool.
3.2 The Challenge of universality and the Realization of uMLIP
While Behler and Parrinello clearly demonstrated the usefulness of MLIP, a major hurdle remained: "universality." Machine learning models, in principle, struggle to predict unknown situations (extrapolation) not included in the training data. Therefore, MLIPs trained on specific elements or atomic environments cannot accurately predict energy and force in different systems, causing simulations to break down. This is essentially the same problem as with classical potentials, requiring recalibration with every change in the system under study. Overcoming this challenge requires a highly expressive model architecture capable of describing diverse atomic environments and a broad dataset necessary for its training. Research in this direction gave rise to the uMLIP trend. In ANI-1, published in 2017, the architecture proposed by Behler and Parrinello was extended to support multi elements, and efficient methods for generating training data were introduced. As a result, MLIPs corresponding to four elements, H, C, N, and O, were constructed, and it was shown that they could be calculated with high accuracy even for systems larger than the training data (transferability) [5].
While conventional MLIPs were limited to a small number of elements, PFP achieved a world-first high level of universality by supporting 45 elements.[6] This leap was made possible by its unique graph neural network architecture and training data that intentionally incorporates even virtual and unstable structures. PFP is truly groundbreaking, overturning the conventional wisdom of MLIPs. Matlantis Corporation, to which the authors belong, provides the computing platform "MATLANTIS™" which makes PFP available.[7] PFP is continuously improved at approximately every six months, and in September 2024, it achieved support for96 elements, encompassing all naturally occurring stable elements. Up to this point, PFP has become a uMLIP that reproduces the accuracy of DFT quickly and over a wide range, but if the error between experiment and calculation can be further reduced, more efficient materials development can be expected. Generally, DFT can improve the accuracy of calculation results by using a high-precision exchange-correlation functional, but this comes with the trade-off of increased computation time. In contrast, once MLIP has completed training, its inference speed remains constant regardless of the specific exchange-correlation functional used to generate the training data. In other words, by training MLIP with more accurate computational results, it becomes possible to obtain more accurate results while maintaining the same inference speed. This approach was embodied in PFP v8, released in June 2025. PFP v8 incorporates an r²SCAN mode, which is trained using the r²SCAN exchange-correlation functional, which is more accurate than the PBE exchange-correlation functional used until now, and it has been confirmed that this can improve the error with experimental values.
As of 2025, the frontier of uMLIP, opened up by PFP, has become a major trend involving academia and industry. Many companies and research institutions have developed their own uMLIPs, and the field as a whole is experiencing rapid development. In response to this boom, a paper on the practical use of uMLIP has been published [8]. With Dr. Takamoto, one of PFP's lead developers, as a co-author, this paper highlights PFP’s role as a leading technology shaping the landscape of this research area.
4. Materials development using uMLIP
So far, we have given an overview of the development history and current status of uMLIP. However, the true value of uMLIP will be realized when it is utilized in practical materials development. In this section, we will introduce specific examples of materials development using PFP.
4.1 Screening of ammonia synthesis catalysts
The development of catalysts for the Haber-Bosch process described above is a good example of how uMLIP can change the development process. Using PFP, researchers identified multiple new catalyst candidates with predicted activities exceeding conventional ones. This was achieved by rapidly screening approximately 2,000 diverse alloy catalysts based on their N adsorption and NN transition state energies [9]. The large scale screening with DFT was difficult in terms of computation time and in terms of elemental species with conventional MLIP, but it was made possible for the first time by the speed and universality of PFP. Thus, PFP is expected to accelerate the discovery of new catalysts in terms of both speed and universality.
4.2 Analysis of polymer thermal decomposition behavior
Understanding thermal decomposition behavior is important for evaluating the heat resistance of polymers and chemical recycling technology. We performed MD simulations of the thermal decomposition of epoxy resins using PFP. As a result, we confirmed that resin decomposition proceeds within a specific temperature range, and that the decomposition mechanism and major products are in good agreement with previous research. [10] While ReaxFF and similar potentials often require tedious parameter adjustments, PFP enables the rapid, experiment-free screening of thermal decomposition properties. This has allowed us to demonstrate an efficient workflow for designing new polymers with excellent heat resistance.
4.3 Evaluation of molecular adsorption properties of MOFs
As of 2025, the most noteworthy materials are likely to be Porous Coordination Polymers (PCPs), also known as Metal-Organic Frameworks (MOFs), which were awarded the Nobel Prize in Chemistry. PCPs/MOFs allow for the design of diverse structures through combinations of metals and organic ligands, and are attracting attention as materials for molecular adsorption and storage. However, due to their vast chemical space, it has been difficult to find structures with desired functions. Shimada et al. utilized PFP to show that the gas adsorption properties of PCPs/MOFs can be explained by the activation energy [11]. In addition, Bonakala et al. have proposed a method to rapidly screen for structures with desired functions from more than 128,000 known MOF structures by combining classical potential and PFP [12]. Until now, the diverse elemental compositions of PCPs/MOFs have made the application of MLIP difficult ; however, the universality of PFP has provided a breakthrough to this challenge.
5. Conclusion and future prospects
This paper provides an overview of how materials development is undergoing a dramatic transformation through the fusion of computational chemistry and AI. Its history began with the "trade-off between accuracy, computation time, and universality." MLIP emerged to overcome this long-standing challenge, achieving both accuracy and speed, and further evolving into uMLIP to gain universality. Now, uMLIP is beginning to fundamentally redefine the materials development process as a game-changer. Of course, even with uMLIP, the computational and spatial scales it can handle are still small compared to phenomena occurring in real materials. To predict macroscopic properties, a materials informatics (MI) approach that integrates experimental and computational data is essential. And uMLIP is expected to play an important role in the MI field as well. It has been reported that uMLIP models, having been trained on diverse atomic environments as foundation models, can be used for transfer learning to specific tasks such as predicting physical properties, it is possible to construct highly accurate predictive models even with small amounts of data [13,14,15]. In other words, uMLIP is not merely a speed-up tool, but is becoming a foundation supporting materials development. On this new foundation, innovative materials essential for future society will be created more rapidly than ever before.
6. References
[1]人類の生存を支えるアンモニア合成② ハーバー・ポッシュ法- 化学工業の幕開け, https://www.jaci.or.jp/gscn/img/page_04/GCS_008-web_v2.pdf (2025年10月28日閲覧).
[2]T. B. Blank et. al., J. Chem. Phys. 1995, 103, 4129.
[3]S. Lorenz, A. Groß and M. Scheffler, Chem. Phys. Lett. 2004, 395, 210.
[4]J. Behler and M. Parrinello, Phys. Rev. Lett. 2007, 98, 146401.
[5]J. S. Smith, O. Isayev and A. E. Roitberg, Chem. Sci. 2017, 8, 3192.
[6]S. Takamoto et. al., Nat. Commun. 2022, 13, 2991.
[7]Matlantis (https://matlantis.com/), software as a service style material discovery tool.
[8]R. Jacobs et. al., Curr. Opin. Solid State Mater. Sci. 2025, 35, 101214.
[9] Catalyst screening for ammonia synthesis catalyst development, https://matlantis.com/en/calculation/ammonia-synthesis-catalyst-screening/ (accessed October 28, 2025)
[10] Thermal decomposition simulation of epoxy molecules, https://matlantis.com/en/calculation/thermal-decomposition-of-epoxy-molecules/ (accessed October 28, 2025)
[11]T. Shimada et. al., Adv. Sci. 2024, 11, 2307417.
[12]S. Bonakala et. al., arXiv preprint arXiv:2509.06719, 2025.
[13]Z. Mao, W. Li, and J. Tan, npj Comput. Mater. 2024, 10, 265.
[14]S. Y. Kim, Y. J. Park, and J. Li, arXiv preprint arXiv:2506.18497, 2025.
[15]T. Shiota, K. Ishihara, and W. Mizukami, Digit. Discov. 2024, 3, 1714.
-




 URL
URL
Copied
Latest Articles
NEW
Learning AI materials simulation to accelerate research at the Fukui Kenichi Memorial Research Center, Kyoto University - Working with ENEOS to design the "best CO2 adsorbent"

Explainer : Why Did the AI Predict That ? Uncovering Atomic-Level Interpretability through PFP Descriptors and Shapley Values

Materials Informatics Explainer computational chemistry

Writing SMILES from scratch
Explainer computational chemistry

Nagoya University × Matlantis Case Study:“Advanced Experiments for Frontier Technologies and Sciences” —A Four-Day Intensive Course That Sparked Experimental Students’ Curiosity Through AI Simulation
Interview computational chemistry

Introduction to Machine Learning Interatomic Potentials (MLIPs): A Game Changer in Materials Simulation

Machine Learning Interatomic Potentials Explainer