In the fields of materials science and pharmaceutical research and development (R&D), significant challenges persist, including the time, cost, and labor associated with experimentation and evaluation, as well as the difficulty of securing skilled personnel. In particular, traditional R&D methods, which often depend on individual experience and intuition, are reaching their limits in simultaneously achieving both development speed and precision.
Materials Informatics (MI) has emerged as a promising solution to these challenges. MI is a data-driven approach that accelerates property prediction and materials discovery by training machine learning models on data obtained from experiments and simulations. A key advantage of this approach is its ability to make inductive inferences from data, rendering it applicable even to complex phenomena where the underlying mechanisms are not fully understood.
This article provides a systematic overview of MI, from its fundamental concepts to the techniques currently in practical use. Furthermore, it addresses the growing convergence of MI and computational chemistry. As a prominent example of this trend, we will also introduce Machine Learning Interatomic Potentials (MLIPs), a technology that leverages machine learning to significantly accelerate MD simulations.
What is Materials Informatics (MI)?
Conventional materials development has historically relied heavily on the experience and intuition of researchers, a process that is often person-dependent, time-consuming, and costly. Materials Informatics (MI) represents a paradigm shift away from this traditional approach. By systematically accumulating and analyzing data with AI technologies, MI transforms materials development into a more sustainable and efficient process. This methodology is being increasingly adopted by numerous research institutions and corporations both domestically and internationally.
MI has the potential to dramatically reduce the number of required experiments by employing machine learning (ML) models to predict material properties and efficiently screen for promising candidate materials. This, in turn, is expected to enhance both the speed and precision of the development cycle.
The research and development of MI is accelerating globally, involving not only top-tier research institutions such as the Massachusetts Institute of Technology (MIT), the National Institute of Advanced Industrial Science and Technology (AIST), and the National Institute for Materials Science (NIMS), but also major chemical companies and IT corporations. Driven by remarkable technological advancements, tangible results are continuously emerging from this field, further expanding its vast potential.
The Two Major Applications of MI: "Prediction" and "Exploration"
The primary applications of MI can be broadly divided into two main categories: "prediction" and "exploration". Understanding the distinct characteristics and appropriate use cases for each is crucial.
(1) Property "Prediction" with ML models
The "prediction" approach involves training ML models on a dataset of known materials. This data pairs input features (e.g., chemical structures, manufacturing conditions) with corresponding measured properties (e.g., hardness, melting point, electrical conductivity). Once trained, the model can be used to predict the properties of novel materials or different manufacturing conditions without the need for physical experiments. This approach leverages vast archives of historical experimental data.
A variety of ML models are employed based on the characteristics of the data, including: Linear models : Linear Regression and its extensions, Ridge and Lasso, Partial Least Squares (PLS). Kernel methods : Support Vector Machine (SVM). Tree-based models : Decision Trees and their advanced ensemble versions, Random Forest and Gradient Boosting Decision Trees (GBDT). Neural Networks.
(2)Efficient "Exploration" with Bayesian Optimization
ML models are not universally accurate; their predictive power is typically high only for data similar to what they were trained on (a process of interpolation). Consequently, when data is scarce or the goal is to discover materials with properties that surpass existing ones, a purely predictive approach has its limitations. This is where the "exploration" approach becomes essential.
The exploration approach utilizes both the predicted mean (the expected value of a property) and the predicted standard deviation (the uncertainty of the prediction) to intelligently select the next experiment to perform. First, it identifies a chemical structure or set of experimental conditions that is most likely to yield a superior property. Then, an experiment is conducted under these conditions, and the new result is added to the training dataset. The model is then retrained, and the cycle repeats. This iterative process enables the efficient discovery of optimal chemical structures and conditions.
This methodology is known as Bayesian Optimization. Within this framework, Gaussian Process Regression (GPR) is frequently used, as it can simultaneously compute both a predicted mean and predicted standard deviation. An alternative method involves training an ensemble of models (e.g., models with different hyperparameters or training data subsets) and calculating the predicted mean and standard deviation from their collective predictions.
While model selection is the primary concern in the "prediction" approach, the "exploration" approach requires careful consideration of not only the model but also the exploration strategy itself. This strategy is governed by the choice of an acquisition function. Numerous acquisition functions have been proposed, with commonly used examples including:Probability of Improvement (PI): Calculates the probability of achieving a result better than the current best.Expected Improvement (EI): Calculates the expected value of the improvement over the current best.Upper Confidence Bound (UCB): A value calculated by adding a multiple of the predicted standard deviation to the predicted mean.
The design of the acquisition function controls the balance between exploitation (leveraging existing knowledge in promising regions) and exploration (investigating unevaluated regions to reduce uncertainty). While a single acquisition function may be used throughout a project, it is also common to switch between different functions as the project progresses.
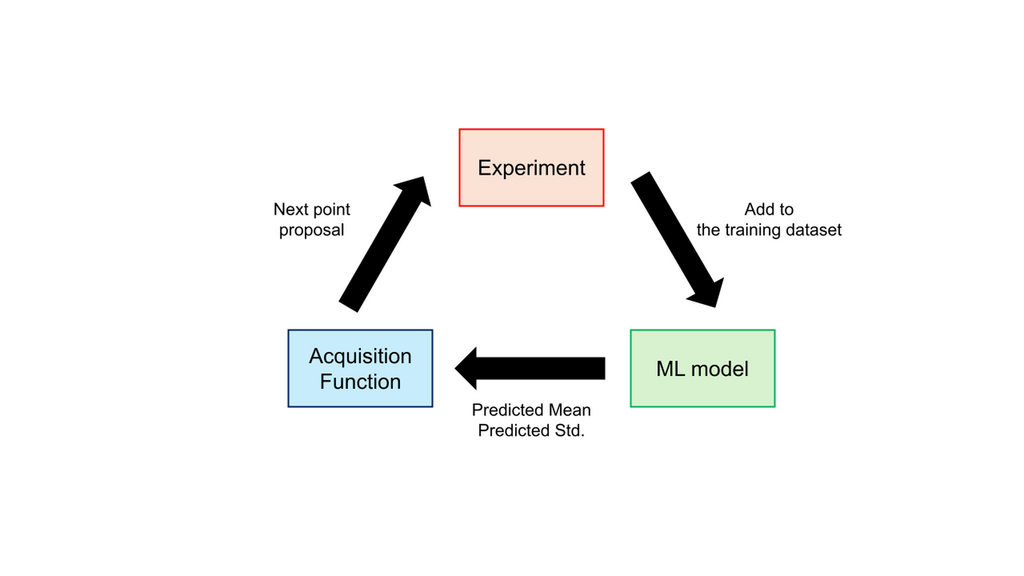
Figure 1. Bayesian optimization flowchart
Representing Chemical Compounds for ML Models: An Overview of Feature Engineering
To apply ML in the chemical domain, information such as chemical structures must first be converted into numerical data that a computer can process. This numerical representation is known as a feature vector or descriptor. This section describes how these features are created.
(1)Feature Engineering Based on Chemical Knowledge
One method for generating features from a compound is to leverage existing chemical knowledge. For organic molecules, this may include descriptors such as molecular weight or the number of substituents. For inorganic materials, features might include the mean and variance of the atomic radii or electronegativity of the constituent atoms. In this approach, a human expert selects the features based on experience and domain knowledge before inputting them into the machine learning model. The primary advantage of using such physically meaningful, knowledge-based features is the ability to achieve stable and robust predictive accuracy even with limited data. However, a significant drawback is that the optimal feature set often varies depending on the class of materials and the target property. Consequently, the feature selection process must be revisited whenever the objective changes.
(2)Automated Feature Extraction with Neural Networks
In recent years, methods that automatically extract features using neural networks have gained considerable attention. Graph Neural Networks (GNNs), in particular, are powerful for this task. GNNs treat molecules and crystals as graphs, where atoms are represented as nodes (or vertices) and chemical bonds are represented as edges. Figure 1 shows the representation of 4-aminophenol as a graph, where each atom is a node with distinct features, and edges connect atoms that share a bond.
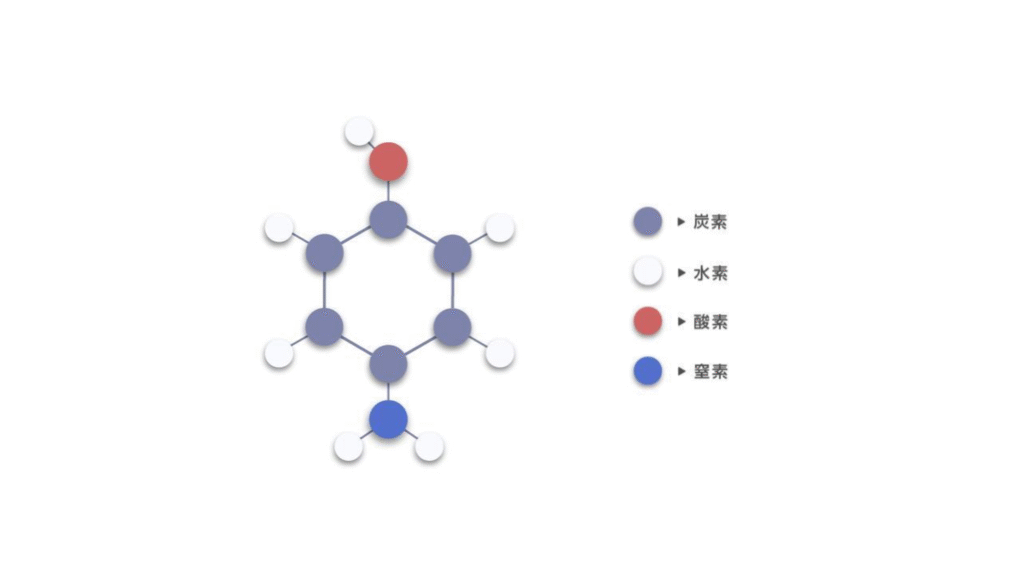
Figure 2. Graph structure of 4-aminophenol
GNNs can automatically learn feature representations that encode information about the local chemical environment, such as the spatial arrangement and bonding relationships between connected atoms, and use these features to predict a target property. This allows the model to directly extract relevant information from the data, leading to high predictive accuracy even in cases where designing effective features manually is difficult. A general requirement, however, is that this approach typically requires a large dataset for training.
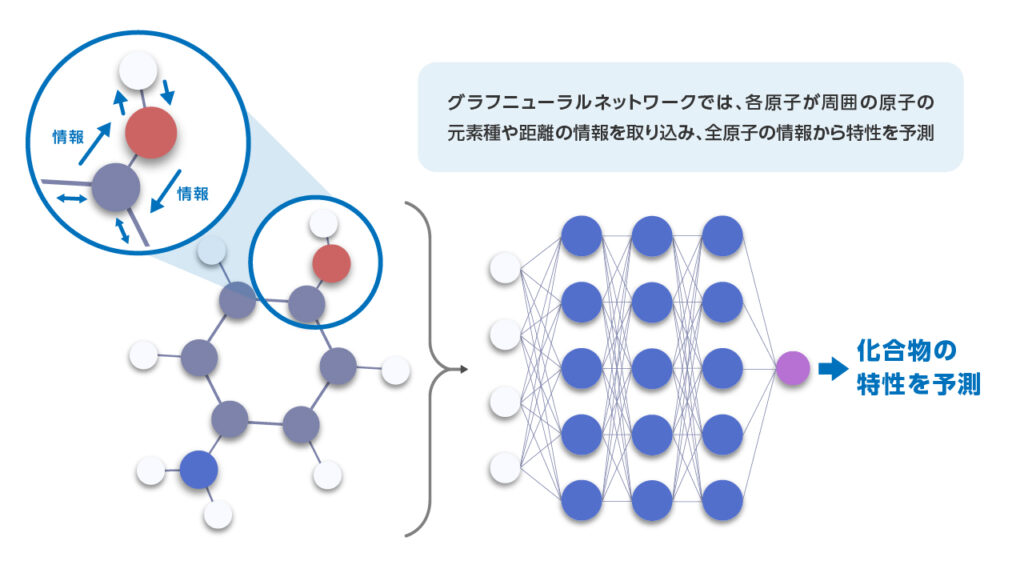
Figure 3. GNN-based prediction of 4-aminophenol properties
The Convergence of MI and Computational Chemistry: An Emerging Paradigm to Overcome Data Scarcity
In MI, training data for ML models is typically acquired through physical experiments. This process can be costly and time-consuming, often resulting in limited datasets for model training. One powerful strategy to address this challenge is the integration of MI with computational chemistry.
This integration is advancing in two primary directions. The first is a conventional approach that aims to improve the accuracy of ML models by using properties calculated from methods like quantum chemistry as input features.
The second, which has gained significant attention in recent years, involves using ML to accelerate computational chemistry itself. The key technology in this area is the Machine Learning Interatomic Potential (MLIP).
To predict material properties with high fidelity, a standard and effective method is to perform Molecular Dynamics (MD) simulations where the interatomic forces are calculated using Density Functional Theory (DFT). However, this DFT-MD approach has faced a significant barrier: the enormous computational time required. MLIP overcomes this by replacing these interatomic interactions with ML models. This allows for a dramatic acceleration of the calculations—by hundreds of thousands of times or more—while maintaining an accuracy comparable to that of DFT.
This breakthrough enables the rapid simulation of diverse structures and conditions that were previously computationally inaccessible, thereby vastly improving the precision of pre-experimental screening. Furthermore, the extensive datasets generated by these high-throughput simulations can then be used as training data for MI models. This creates a powerful synergy that directly addresses the foundational problem of data scarcity, making it a highly promising approach that significantly expands the predictive scope (the interpolation domain) of MI.
Conclusion and Future Perspectives
This article has provided an overview of Materials Informatics (MI), from its fundamental concepts to its advanced applications. MI is not merely the introduction of a new tool; it represents a fundamental paradigm shift in the methodology of research and development, transforming it from a traditional approach based on experience and intuition to a data-driven one.
The core of MI lies in its use of AI and machine learning to enable the "prediction" of material properties and the efficient "exploration" for optimal conditions. While data scarcity remains a key challenge, MLIPs offer a promising solution. By enabling high-speed, high-accuracy simulations, MLIPs make it possible to predict material properties even in the absence of any initial experimental data.
Looking ahead, the challenge of data availability is expected to be further addressed by emerging technologies. These include the automation of experiments through robotics and the use of Large Language Models (LLMs), such as ChatGPT, to convert unstructured text from sources like scientific literature and laboratory notebooks into structured data. It is anticipated that these advancements will resolve data bottlenecks and further accelerate the pace of materials discovery driven by MI.
-




 URL
URL
Copied
Latest Articles
NEW
Learning AI materials simulation to accelerate research at the Fukui Kenichi Memorial Research Center, Kyoto University - Working with ENEOS to design the "best CO2 adsorbent"

NEW
Explainer : Why Did the AI Predict That ? Uncovering Atomic-Level Interpretability through PFP Descriptors and Shapley Values

Materials Informatics Explainer computational chemistry

Writing SMILES from scratch
Explainer computational chemistry

Nagoya University × Matlantis Case Study:“Advanced Experiments for Frontier Technologies and Sciences” —A Four-Day Intensive Course That Sparked Experimental Students’ Curiosity Through AI Simulation
Interview computational chemistry

Introduction to Machine Learning Interatomic Potentials (MLIPs): A Game Changer in Materials Simulation

Machine Learning Interatomic Potentials Explainer