エポキシ分子の熱分解シミュレーション
テーマ概要
エポキシ樹脂の熱分解反応は、プラスチックリサイクルのプロセス設計において重要です。通常、ポリマー分子の熱分解は高温で進行し、様々な小分子が生成されます。
このような反応過程を扱う方法として、反応力場(ReaxFF)を用いた古典分子動力学(MD)シミュレーションが広く用いられています。これにより、分解反応を詳細に解析することが可能です。
一方、ReaxFFは力場の作成が難しいという課題があります。そこで、学習済みの汎用ポテンシャルであるPFPを用いて同様の計算を行い、結果の検証を行いました。
計算モデルと計算方法
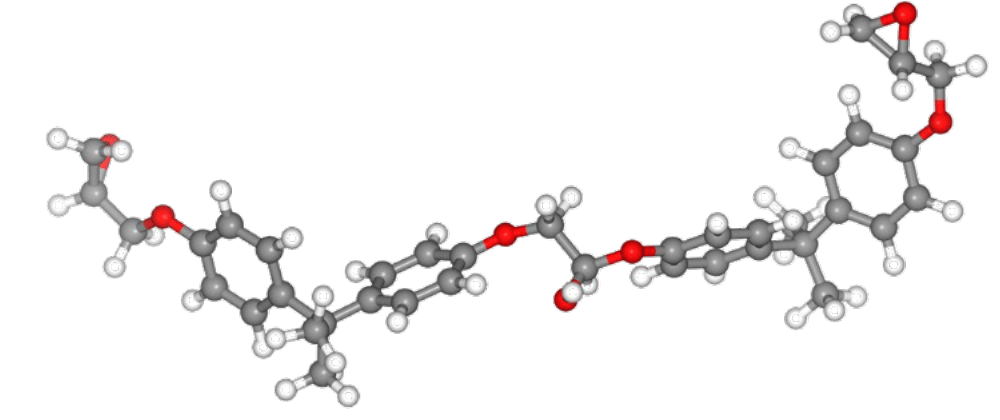
計算にはエポキシ樹脂のモノマー(図1)15分子からなる構造モデルを用いました(図2参照)。NPTアンサンブルによる平衡化を行った結果、系の密度は0.95 g/cm³でした。この値は実験、および、ReaxFFによる先行研究の結果と一致しています。
その後、NVTアンサンブルを用いて温度を上昇させるシミュレーションを行い、熱分解反応に対する温度条件の影響を評価しました。
計算結果と展望
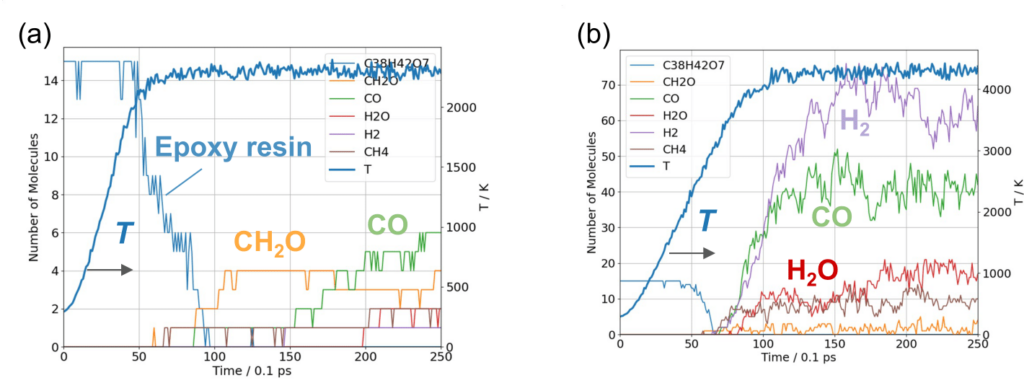
図3に経過時間に対するセル内の分子の個数を示します。最終温度Tend=2300 Kの結果(図3(a))から、2000 K付近からエポキシ樹脂の分子数が減少し、その後に小分子が生成することが分かります。これは、まずエーテル結合の解離が起こり、その後に熱分解が起こることを示しています。小分子の生成は、CH2O、CO、CH4の順に起こっています。
より高温のTend=4300 Kの場合、2000 K以上においてH2、CO、H2Oが生成しています(図3(b))。Tend=2300 Kの結果に対して、より小さな分子にまで分解がすすむことが分かります。
この結果は先行研究と一致し、PFPが有機化学反応を精度良く計算できることを示しています。
計算条件

参考文献
[1] Diao, Z.; Zhao, Y.; Chen, B.; Duan, C.; Song, S. Journal of Analytical and Applied Pyrolysis 2013, 104, 618-624タグ
本事例の公開日:2024.04.24
-




 URLを
URLを
コピーしました