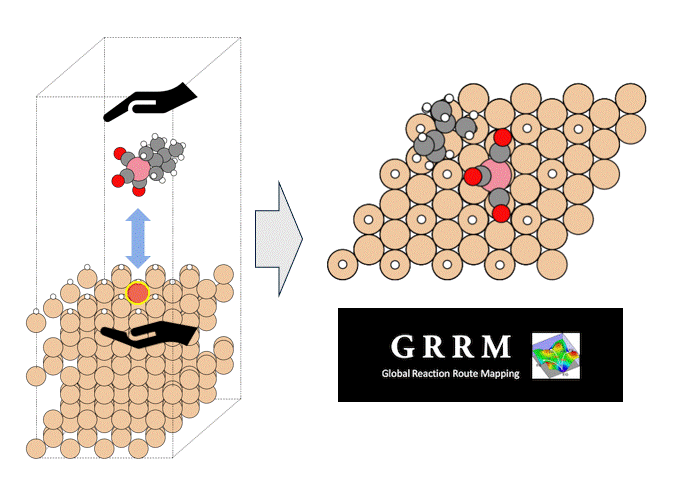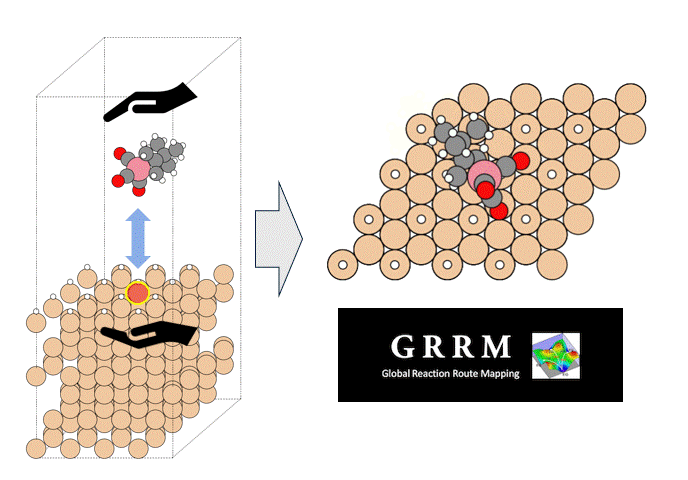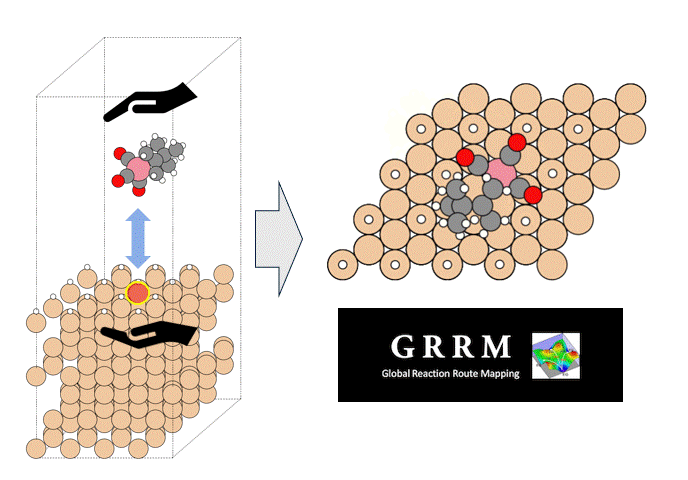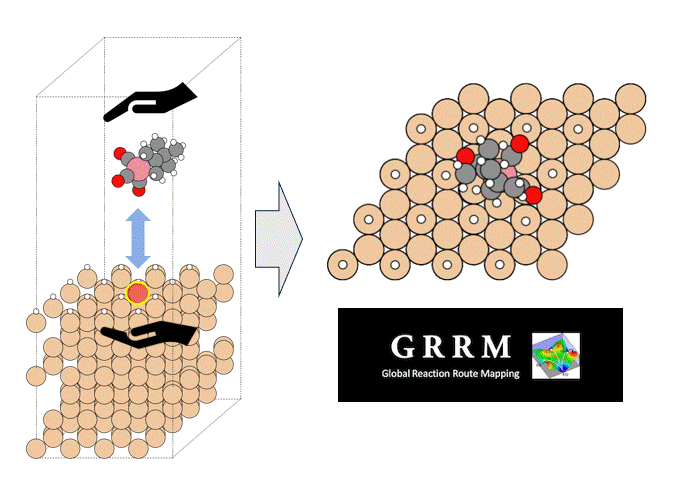
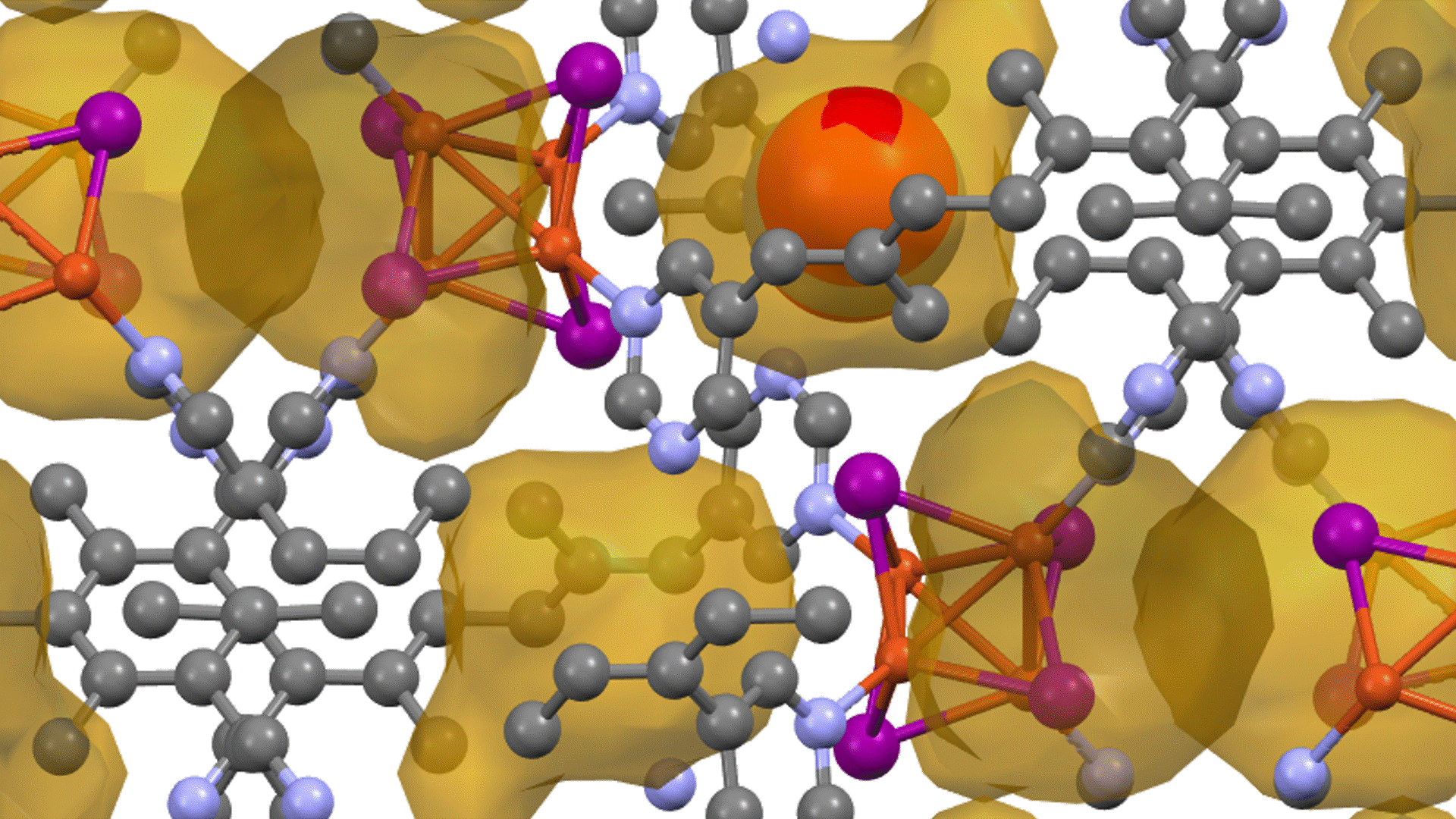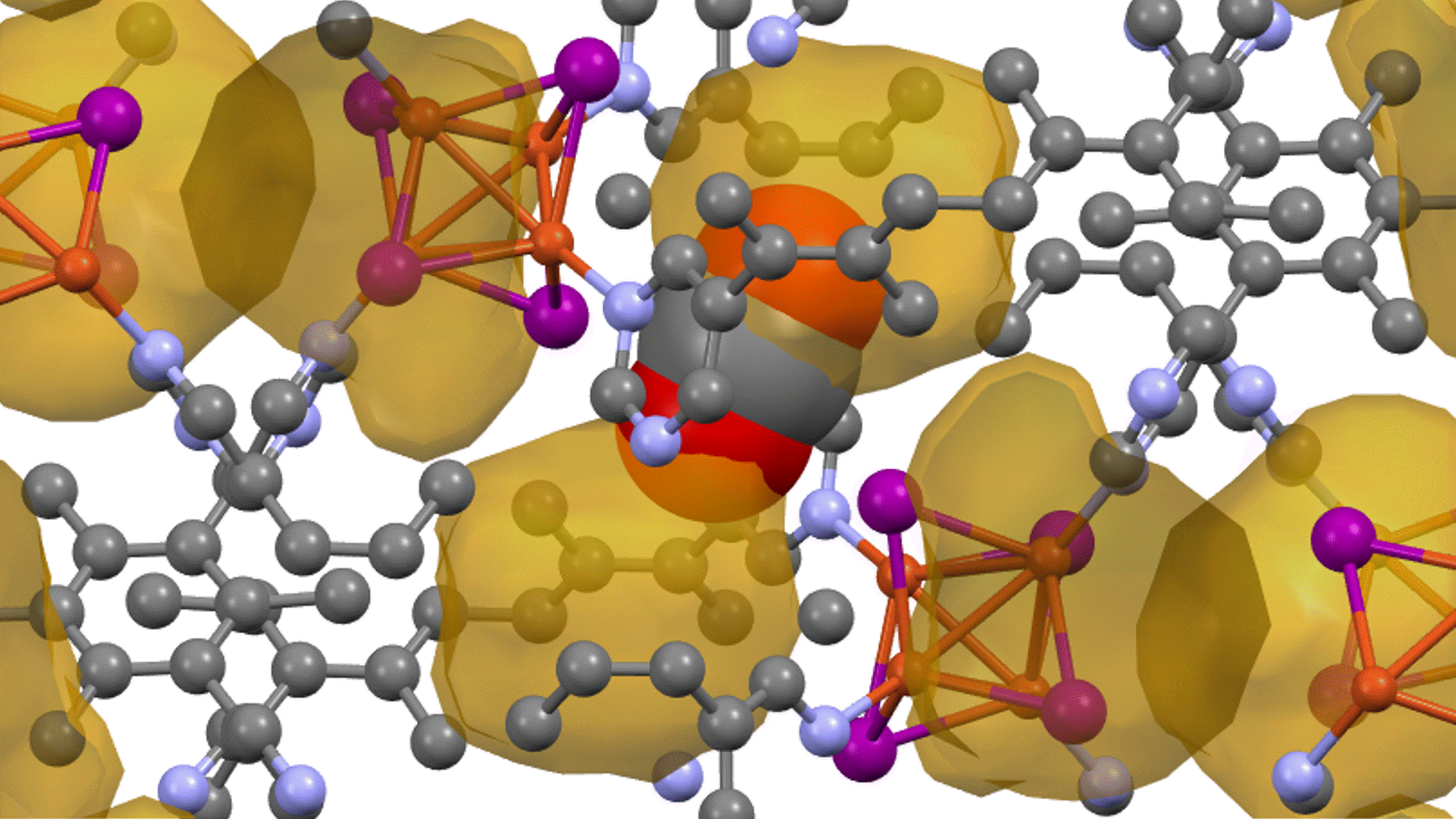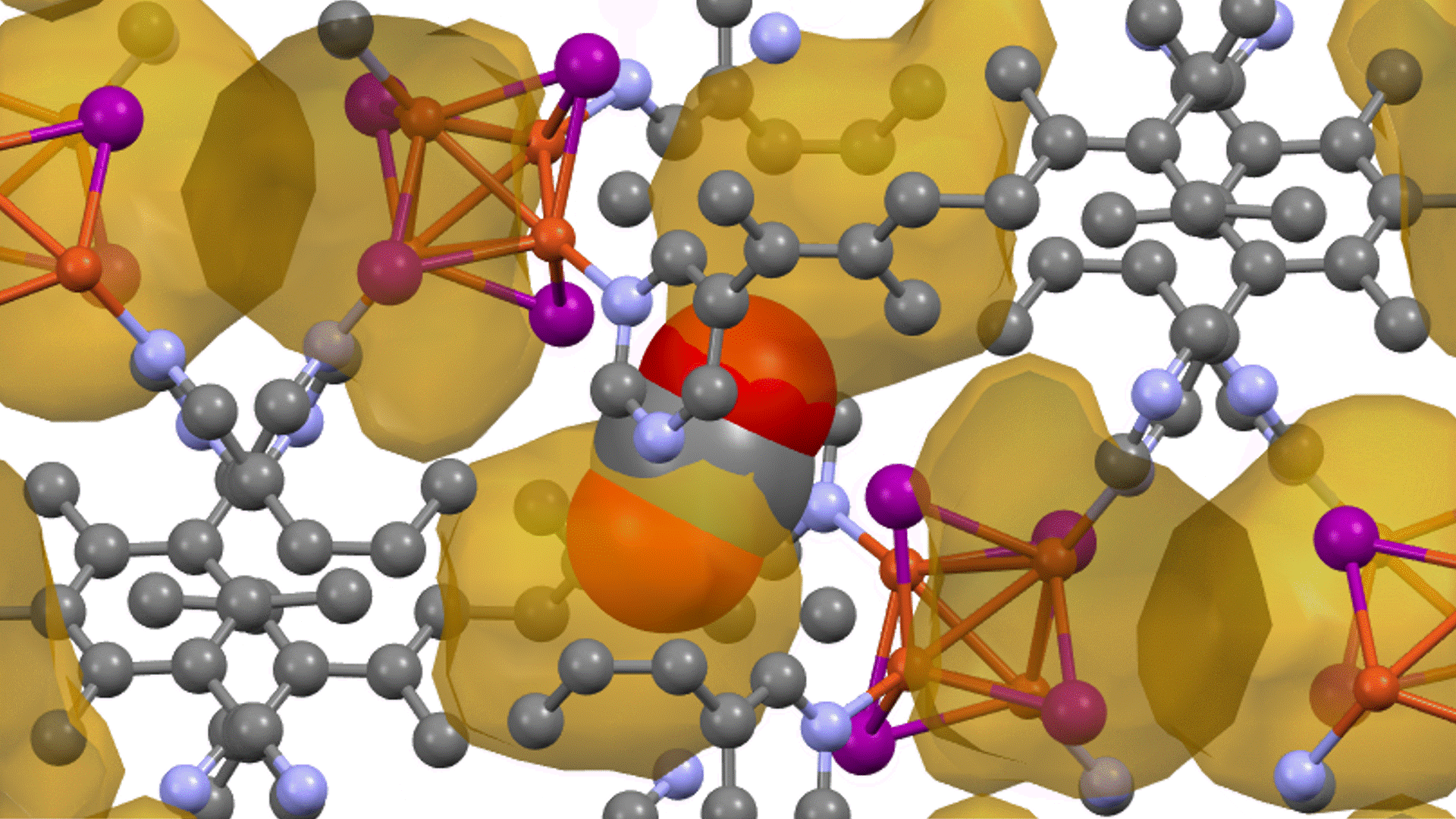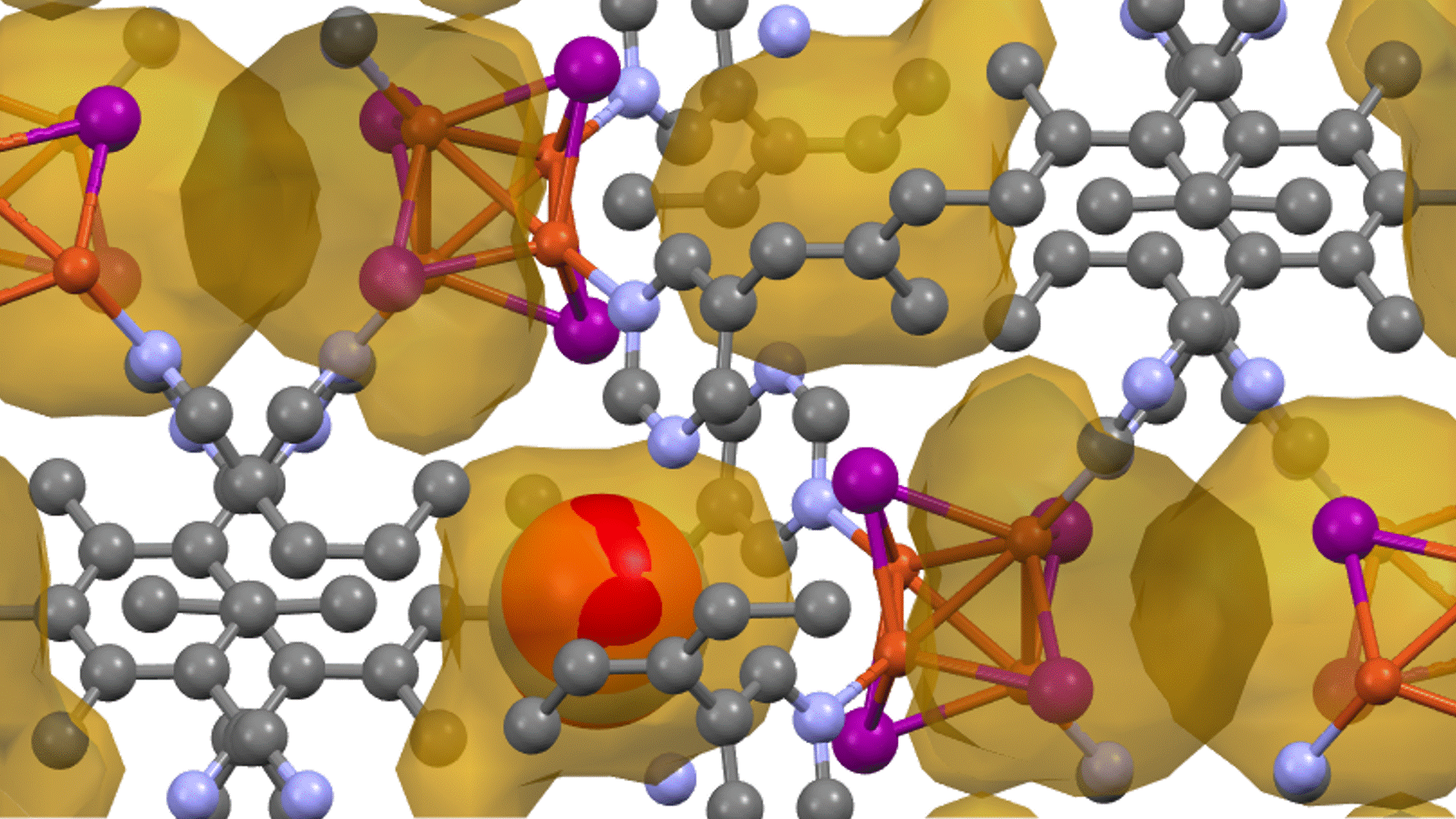
硫化物固体電解質中のLi拡散
テーマ概要
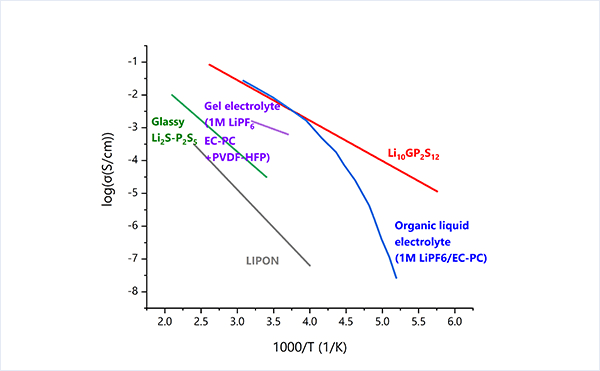
イオンを通す固体電解質は古くから知られていますが、過去10年程で目覚ましい発展を遂げてきました。特に硫化物系の固体電解質はイオン伝導度が飛躍的に向上し、全固体のリチウムイオン電池への応用が期待されています。
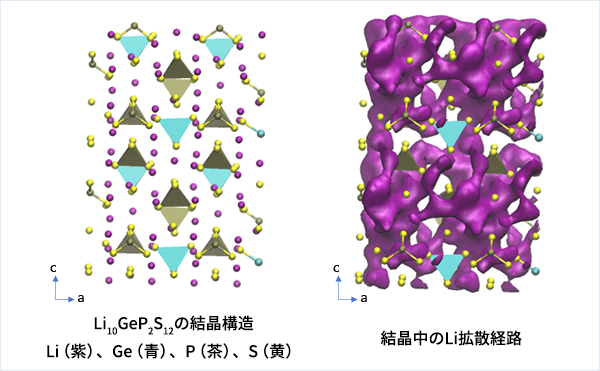
Li10GeP2S12(LGPS)はその中でも最も高いLi-ion伝導度を示す部類に属し、その特徴的な結晶構造が重要なことが分かってきています。
今回はMatlantisを使ってLGPS中でのリチウムイオン拡散係数を分子動力学計算により求めてみます。
本計算事例のサンプルスクリプトはmatlantis-contrib上で公開されています。
計算モデルと計算方法
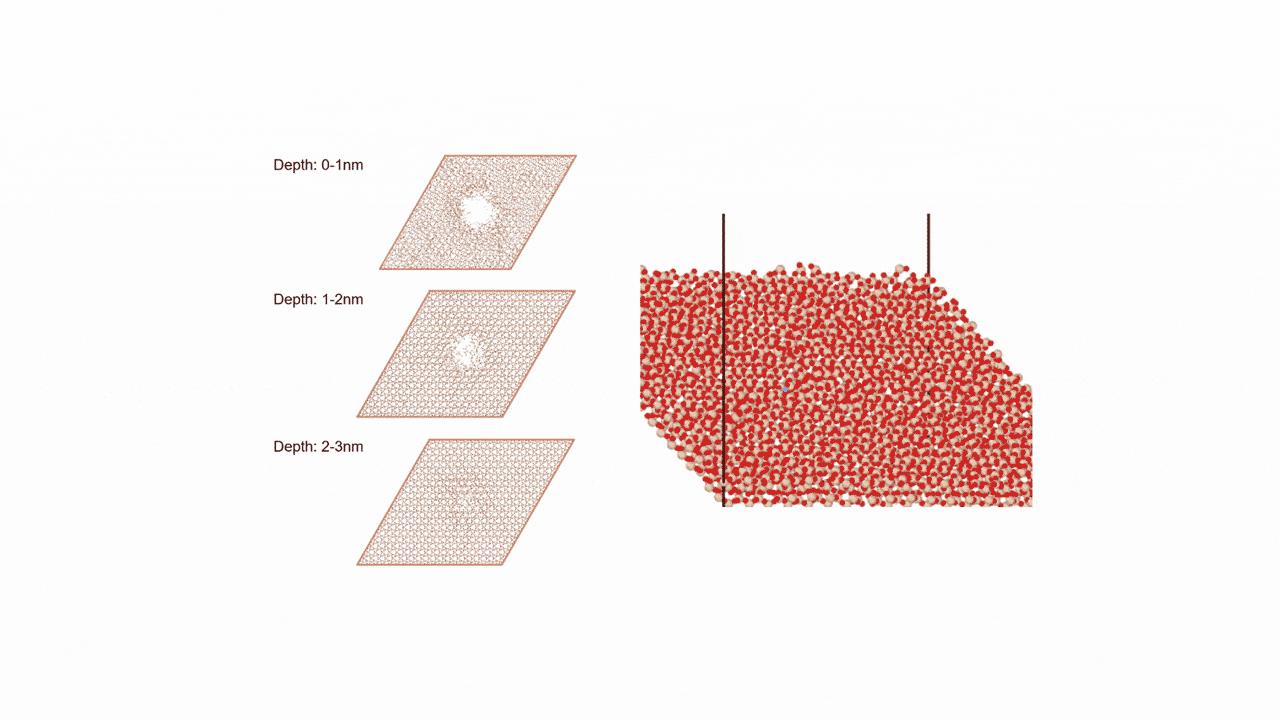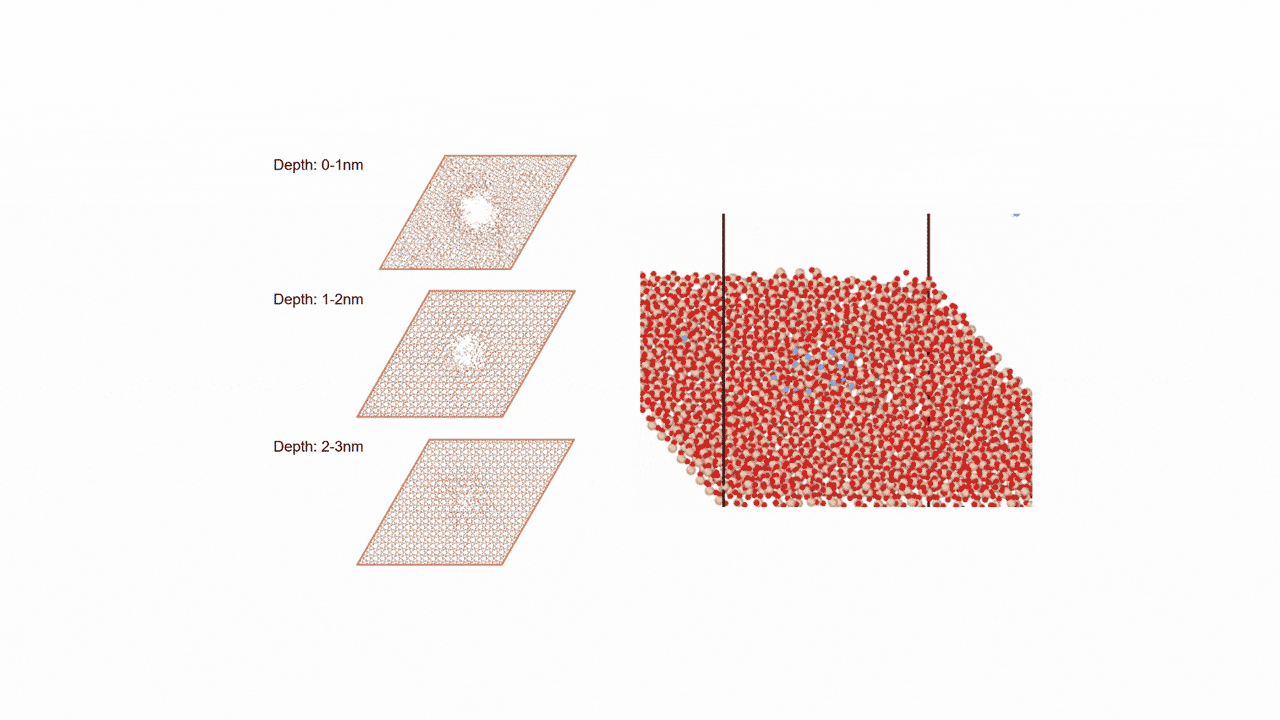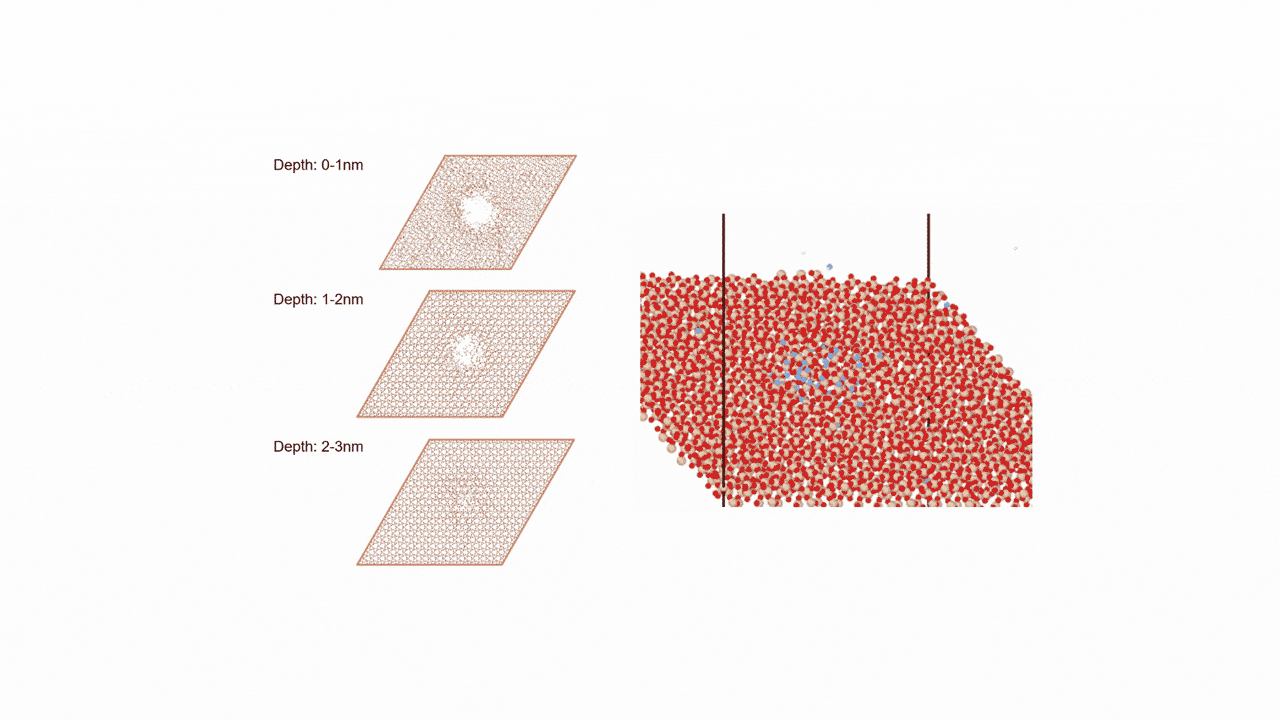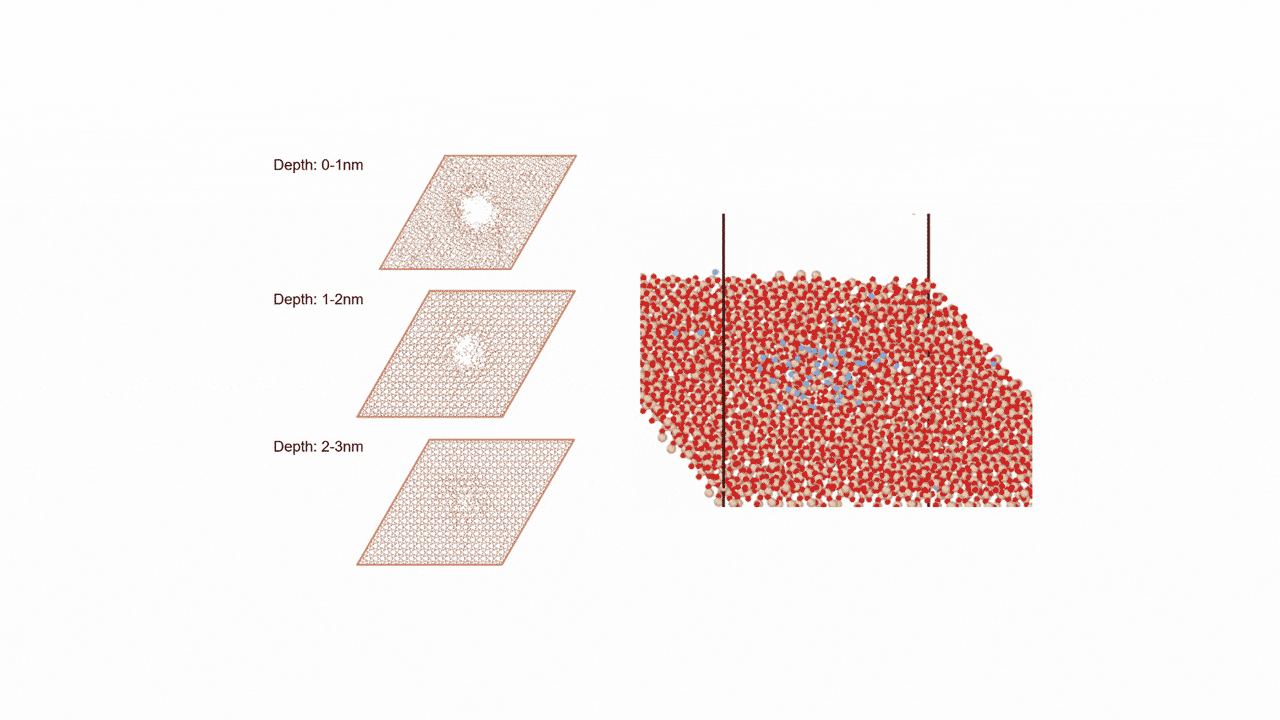
ここではLGPSの結晶構造の最適化を行い、Liの拡散係数を算出します。
LGPSの結晶構造は原論文[1]もしくはMaterials Project[2]から取得可能です。取得した構造をベースに、構造最適化により再現性を確認したうえでリチウムイオンの挙動を分子動力学で解析していきます。
構造最適化が完了した構造に対しMDシミュレーションを実行していきます。今回はNVT ensembleを採用するためLangevin dynamicsを実行します。
計算結果と展望
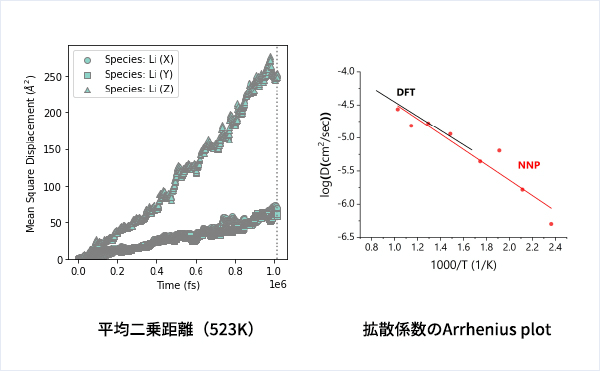
MDの結果からリチウムイオンの平均二乗距離をプロットしてみたところ、Z方向のMSDが突出して伸びていることが確認できます。LGPSではc軸方向(=z軸方向)への拡散が最も顕著で、それと垂直方向であるXY平面上での拡散は限定されていることが知られています。MSDのプロットはそのような特徴を明確に表しています。
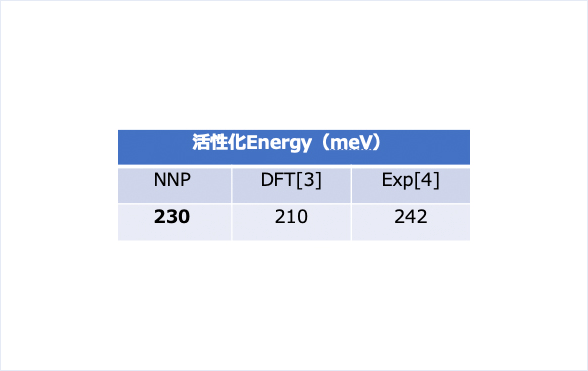
得られたMSDから拡散係数を求めプロットしてみました。文献のDFTの値と比較しても非常によく合っており、整合性の高い結果が得られています。活性化Energyに関しても文献のDFTの値、および実験値とよく一致しています。[3-4]
従来のDFTでは非常に計算コストが高いため高温で行う必要があったMDシミュレーションもNNPを用いるとより低温側で実行することが可能になり、実験データとの整合性も高いことが確認出来ました。
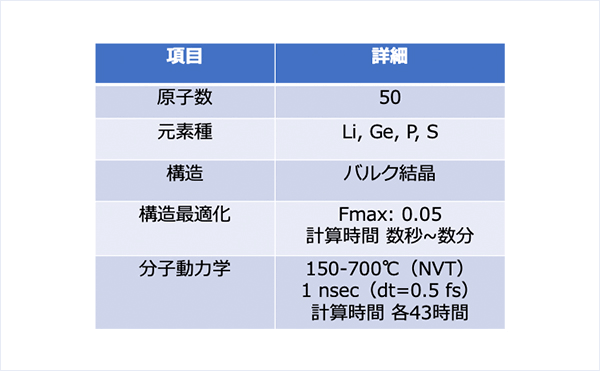
計算条件
References
[1] N. Kamaya, et. al., Nature Mater 10, 682–686 (2011). https://www.nature.com/articles/nmat3066 [2] https://materialsproject.org/ [3] Mo et al. Chem.Mater. (2012) 24, 15-17 https://pubs.acs.org/doi/10.1021/cm203303y [4] Y. Kato, et. al. Nat. Energy 1, 16030. https://www.nature.com/articles/nenergy201630タグ
本事例の公開日:2022.01.21
-




 URLを
URLを
コピーしました