High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
Overview
In drug discovery, the accurate prediction of protein–ligand binding affinity during the lead optimization stage is critical to the success of compound selection. However, high-accuracy interaction calculations based on quantum chemistry typically require tens of hours per structure, making them impractical for large-scale compound libraries. In this study, we evaluated the binding energies of Cyclin-Dependent Kinase 2 (CDK2)—a well-known target for cancer therapy—and six types of inhibitors using Matlantis. The results demonstrated that Matlantis achieved a computational speedup of approximately 34,000 times compared to the Fragment Molecular Orbital (FMO) method while maintaining equivalent or superior accuracy (R2 = 0.95).
Background
Quantum chemical calculations are widely used to obtain a deeper understanding of protein–ligand interactions in biological systems and to support the rational optimization of lead compounds and prediction of binding affinity. Recently, approaches that account for protein flexibility and structural fluctuations have become increasingly important.
For instance, research by Takaba et al. (J. Comput. Chem. 2022) [1] demonstrated that the DA-FMO (Dynamic Averaging FMO) method—which dynamically averages energies across a structural ensemble obtained from MD simulations—significantly improves the correlation between calculated binding energies and experimental values compared to using a single crystal structure (improving R2 from 0.75 to a higher correlation).
However, while quantum chemical methods such as FMO can provide highly accurate energy evaluations, calculations for systems containing tens of thousands of atoms (including protein, ligand, and solvent) often require tens of hours per structure. Consequently, performing FMO calculations on a large number of snapshots requires enormous computational resources and time, posing a significant barrier to practical application.
Target System
Cyclin-Dependent Kinase 2 (CDK2) belongs to the serine/threonine kinase family and is a key protein regulating transitions between phases of the cell cycle. Abnormal CDK2 activity is closely linked to tumor growth, making it a prominent target for anticancer drug development.
In this case study, we examined six CDK2 inhibitors (CS12, CS9, CS242, CS245, CS246, and CS262) whose binding modes have been elucidated by X-ray crystallography. All of these inhibitors are electrically neutral under physiological conditions, and their experimental binding free energies (Gexp) range from −4.88 to −10.00 kcal/mol.
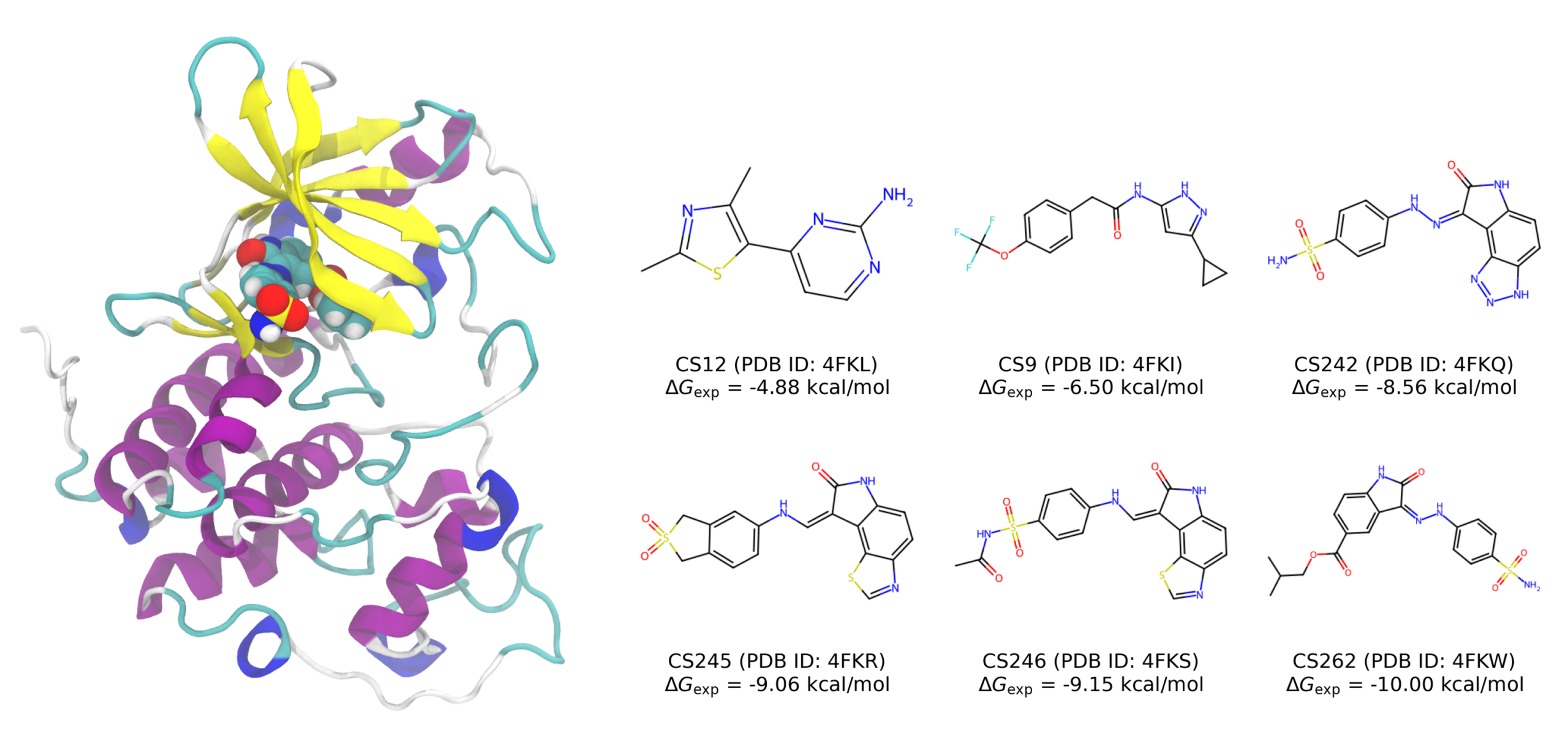
Computational Methods
Preparation of Structural Ensemble
We used MD simulation trajectories conducted by Takaba et al., which is registered in the FMODB [1, 2]. The system consists of roughly 10,000 atoms, including the protein, ligand, and solvent. For each protein–ligand structure, snapshots were extracted every 2 ns from the period after 12 ns in five 50 ns simulations, yielding a total of 100 structures for binding energy calculations of each ligand.
Estimation of Binding Energy
Binding energy was calculated using the following supramolecular approach based on the energy difference between the complex, the protein, and the ligand:
For each ligand, binding energies were calculated for 100 snapshots (600 structures in total), and the mean and variance of these values were then computed.
Methods for Comparison
We evaluated the binding energy using the following two methods, and their correlations with the experimental free energies (ΔGexp) were compared.
Table 1: Methods compared
| FMO (DA-FMO) | Matlantis (PFP) | |
| Level of Theory | FMO-MP2/6-31G* | PFPv8 (R2SCAN + D3) |
| Energy Evaluation | Inter-Fragment Interaction Energy (IFIE) | Supramolecular binding energy (ΔEbind) |
| Software | ABINIT-MP | Matlantis |
Table 2: The number of atoms in the calculated systems
| Protein | Ligand | Water molecule | Total | |
| CS12 (PDB: 4FKL) | 4848 | 24 | 6897 ~ 7524 | 11769 ~ 12396 |
| CS9 (PDB: 4FKI) | 4848 | 37 | 6861 ~ 7452 | 11746 ~ 12337 |
| CS242 (PDB: 4FKQ) | 4848 | 36 | 6963 ~ 7482 | 11847 ~ 12366 |
| CS245 (PDB: 4FKR) | 4848 | 39 | 6873 ~ 7533 | 11760 ~ 12420 |
| CS246 (PDB:4FKS) | 4848 | 42 | 6882 ~ 7479 | 11772 ~ 12369 |
| CS246 (PDB:4FKS) | 4848 | 49 | 6879 ~ 7467 | 11776 ~ 12364 |
Results
Prediction Accuracy of Binding Energy (ΔEbindvs ΔGexp)
The correlation between the calculated binding energy (ΔEbind) and the experimental binding free energy (ΔGexp) was evaluated for each inhibitor. The results are shown in Figure 2 and Table 3.
Table 3: Comparison of Prediction Accuracy
| Method | R² | RMSE (kcal/mol) |
| FMO (DA-FMO) | 0.91 | 0.52 |
| PFPv8 (R2SCAN + D3) | 0.95 | 0.38 |
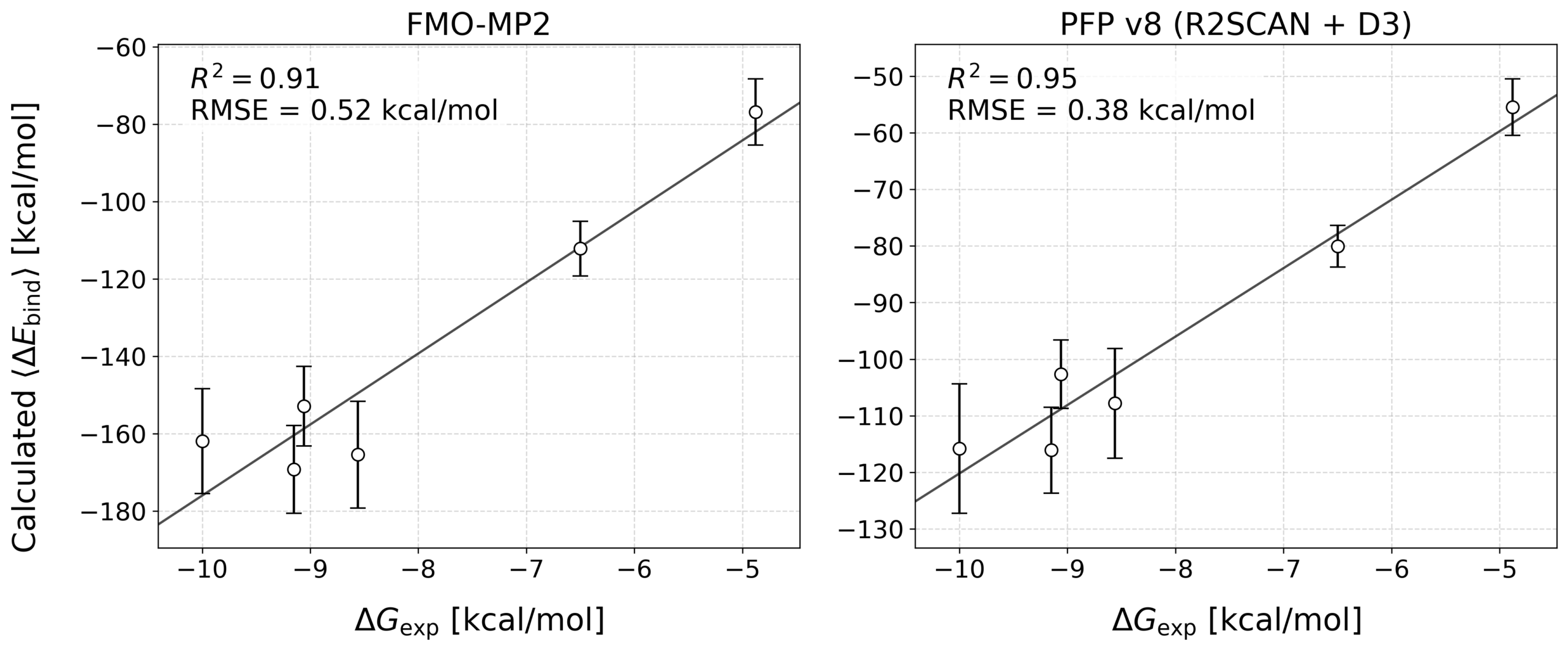
Matlantis (PFP) showed a smaller error (RMSE) and an excellent correlation with the experimental binding free energies compared with FMO. In other words, this result confirms that Matlantis can evaluate protein–ligand interactions with accuracy comparable to traditional quantum chemistry methods.
Comparison of Computational Speed
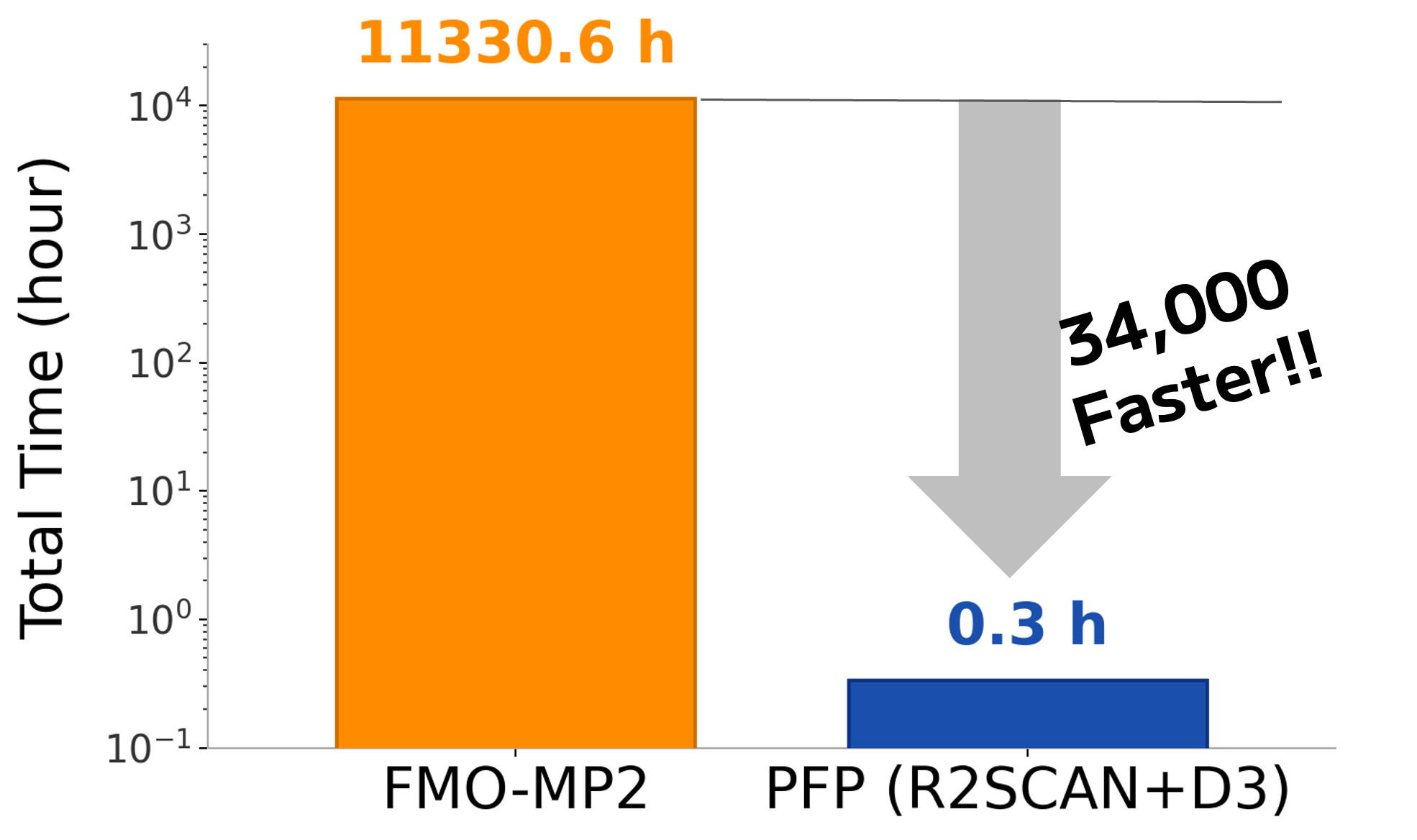
Next, we compared the required computation time. For the total of 600 structures, FMO required 11,330 hours (approximately 472 days) of computation(*), whereas Matlantis (PFP) completed the calculation in only 20 minutes. In other words, Matlantis (PFP) enabled binding energy evaluation at approximately 34,000 times higher speed.
When extrapolated to a screening scale, evaluating 1,000 compounds × 100 snapshots (100,000 structures in total) would require approximately 5.2 years with FMO, whereas Matlantis would complete the same calculation in about 56 hours. Since the prediction accuracy is comparable to or better than FMO, this demonstrates that interaction evaluation at a quantum chemistry level can now be applied even to large-scale compound exploration that had previously been infeasible because of computational cost constraints.
* Computational time for FMO is obtained from logfile deposited on FMODB[1, 2]
Conclusion
Averaging interaction energies over ensemble structures obtained from MD simulations is highly effective for improving the accuracy of binding affinity prediction. However, applying conventional quantum chemical calculations to screening applications has been a major challenge because of the enormous computational resources and time required.
In this case study, we demonstrated that by using Matlantis (PFP), it is possible to dramatically reduce computation time while maintaining prediction accuracy at the level of quantum chemical calculations. This makes it feasible to efficiently evaluate interactions across large chemical spaces that were previously inaccessible due to computational cost. As a result, promising compound discovery in drug design and agrochemical design can be significantly accelerated.
References
[2] FMO Database (FMODB): https://drugdesign.riken.jp/FMODB/
tag
Publication date of this case study: 2026.05.20
-




 URL
URL
Copied
what's new
Data-Driven Method for Discovering Low Adsorption Energy Molecules on Si Surfaces
ScreeningMaterials InformaticsSemiconductors
Calculation and Verification of Methanol Synthesis Catalysts
Methanol synthesis catalystsLarge-scale screening
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis

SiO₂ Dry Etching Simulation Using LightPFP
Semiconductor