A wide variety of materials can be simulated using Matlantis, a machine learning–based atomistic simulation platform that enables fast and accurate prediction of atomic structures and properties. Among these materials, Metal-Organic Frameworks (MOFs) are particularly important materials with a broad range of applications, including catalysis and CO2 storage. Discovered in the 1990s, these materials are already being industrialized and are gaining global attention as essential components for a sustainable society. In fact, the 2025 Nobel Prize in Chemistry was awarded to the three researchers who discovered this rich family of materials. This article reports the results of calculations performed on the MOFSimBench benchmark, released in July 2025, to verify the predictive performance of Matlantis for MOF materials. It also includes a comparison with other open-source machine learning potentials.
MOFSimBench Overview and Calculation Conditions
MOFSimBench consists of a diverse set of 100 structures collected from existing MOF databases such as QMOF, MOSAEC-DB, IZA, and CURATED-COF, with an emphasis on diversity, as well as structures from the CoRE MOF and GoldDAC databases [1]. The benchmark consists of five tasks: structure optimization, molecular dynamics, bulk modulus, specific heat, and host-guest interaction. The performance of various open-source machine learning interatomic potentials (MLIPs) is evaluated against these tasks. Additionally, the energy prediction accuracy for the QMOF database and the calculation speed of each model are also reported as supplementary information. The reference ("correct") data is calculated using DFT with the PBE functional. The MLIPs compared include prominent models such as MACE, Orb, SevenNet, MatterSim, and eSEN-OAM. To account for dispersion forces in the predictions of these models, the torch-dftd package, released as open-source by Preferred Networks, Inc., is used [2].
In this article, we performed validation using MOFSimBench for our model. We also discuss calculation speed based on the results from reference [1] and benchmarks previously obtained by our company. The model used is the latest version available on Matlantis as of October 2025, PFP v8.0.0, in the PBE PLUS D3 mode, which includes dispersion correction. PFP is accelerated by its proprietary inference engine, PFVM [3]. Furthermore, we also tested a small model of Meta's UMA (Universal Models for Atoms), which was not evaluated in MOFSimBench (model name: uma-s-1p1) [4]. UMA requires switching parameters called "task_name" depending on the material to be calculated; here, we set "odac" as the task_name. An NVIDIA Tesla T4 GPU was used for the uma-s-1p1 calculations. The prediction accuracies for all other models were cited from the values reported in the references.
(1) Energy Prediction
Figure 1 shows a comparison of PFP's prediction results for approximately 20,000 structures in the QMOF database [5] and the energy prediction performance of various MLIP models. As is clear from the parity plot on the left, PFP shows excellent agreement with the DFT results, with a Mean Absolute Error (MAE) of 0.006 eV/atom. This result is superior to models such as eSEN-OAM and equiformerV2.
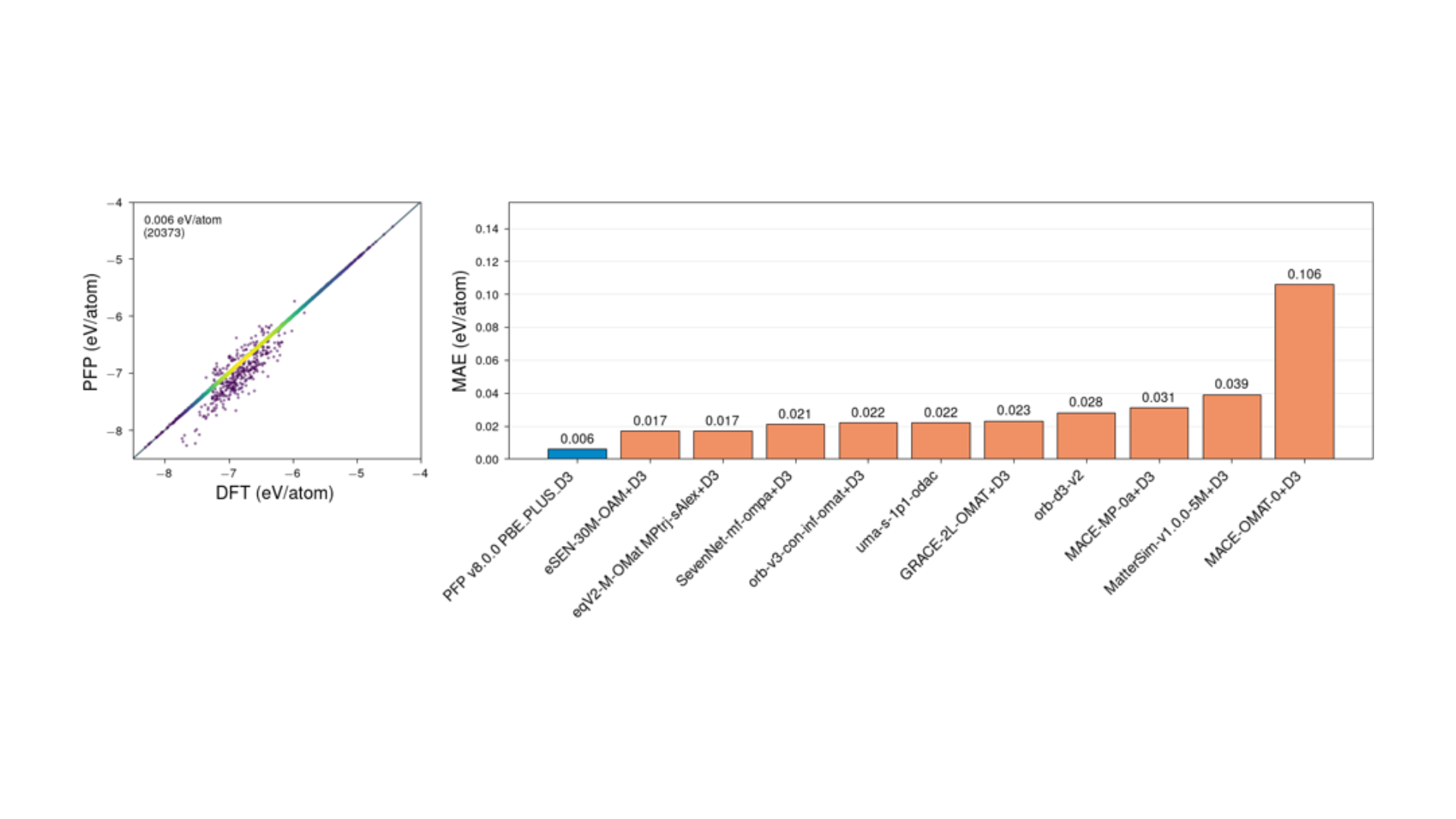
Figure 1. Evaluation of energy prediction accuracy for QMOF. Left: Parity plot of PFP vs. DFT. Right: Comparison of prediction error (MAE) by model.
(2) Structure Optimization
In the structure optimization task, MLIP was used to optimize 100 structures (83 MOF structures, 7 COF structures, and 10 zeolite structures), and the volume change rate compared to the optimized structures obtained by DFT was evaluated. The left side of Figure 2 shows a violin plot of the volume change rate between PFP and DFT for the 100 optimized structures. The gray area corresponds to the ±10% region. As can be seen, PFP achieved 92 of the 100 structures within this region. The bar graph on the right side of Figure 3 compares the number of structures within the ±10% region for each model. As can be seen from this figure, PFP achieved the highest level of performance among these MLIPs. Next, orb-v3-omat+D3, eSEN-OAM, and uma-s-1p1 showed excellent performance.
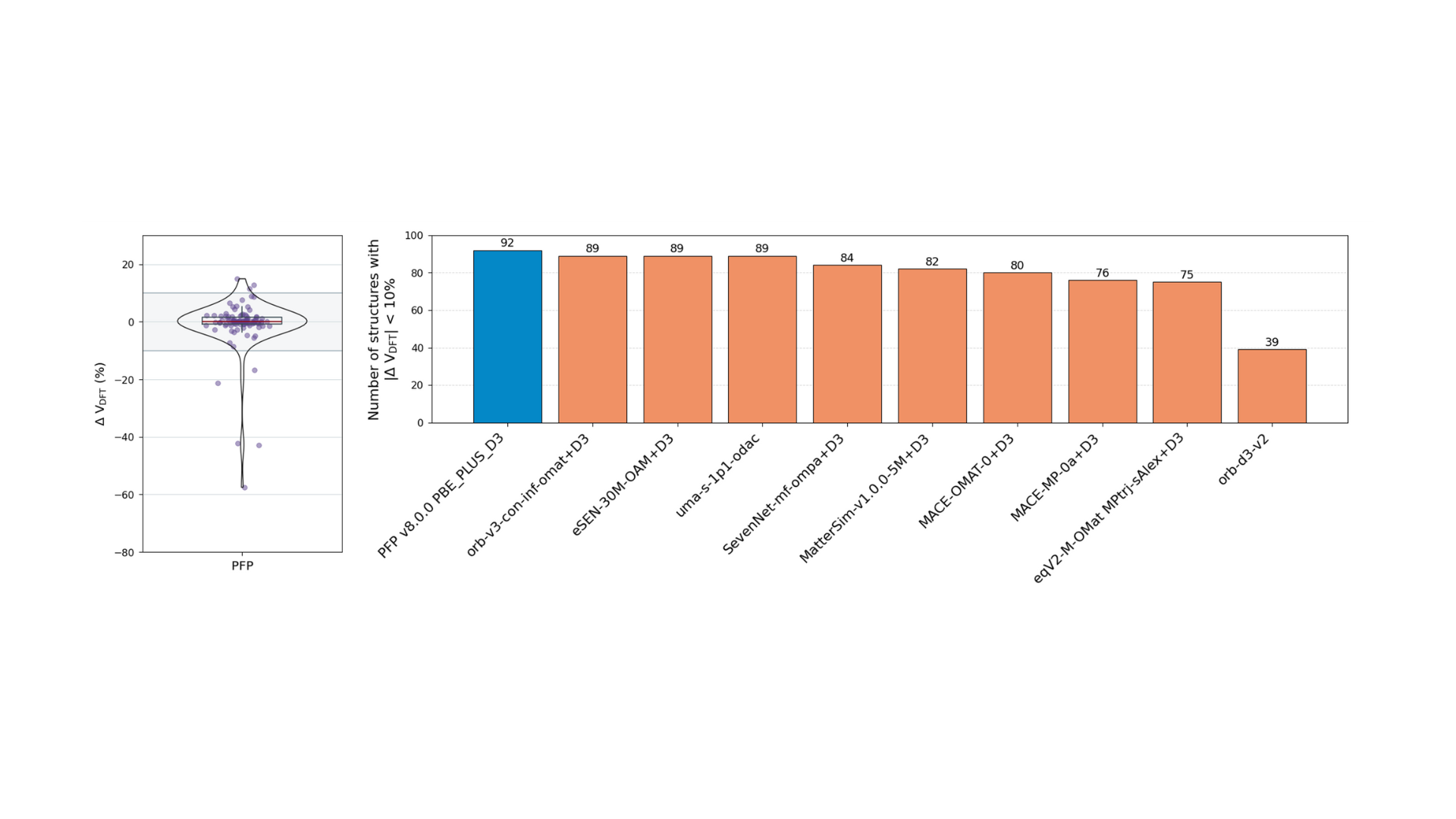
Figure 2. Evaluation results for the structure optimization task. Left: Violin plot of volume change (ΔVDFT= 1 – V / VDFT= 1 – V / VDFT). Right: Comparison of the number of structures where |ΔVDFT| < 10%.
(3) Molecular Dynamics Calculation
Stability was also evaluated through molecular dynamics calculations. Similar to the structure optimization task, calculations were performed on 100 structures. After equilibrating the structures through optimization and NVT calculations, an NPT simulation was run for 50 ps at 300K and 1 bar. The volume change between the initial and final structures was then evaluated. As in the previous task, the bar graph on the right compares the number of structures with an absolute volume change rate of less than 10% for each model. Models such as eSEN-OAM, PFP, and orb-v3-omat+D3 were top performers. Note that the molecular dynamics task was not evaluated for uma-s-1p1 as sufficient calculation speed could not be achieved with the Tesla T4 GPU.
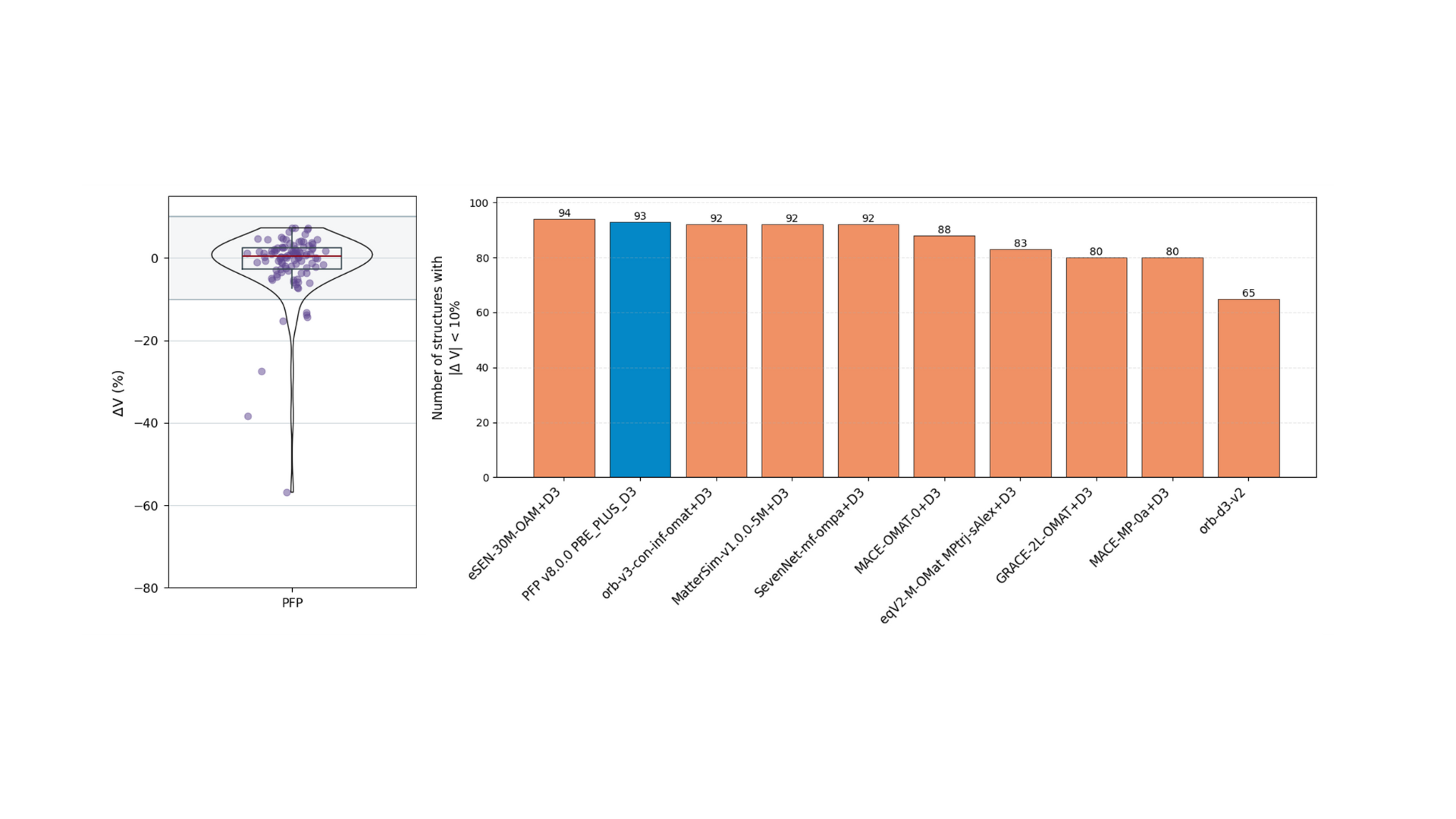
Figure 3. Evaluation results for the molecular dynamics calculation task. Left: Violin plot of volume change (ΔV= 1 – Vfin/ Vini). Right: Comparison of the number of structures where |ΔV| < 10%.
(4) Bulk Modulus
In MOFSimBench, the predictive performance for bulk properties of MOF materials is evaluated by the bulk modulus and specific heat. Figure 4 shows the prediction results for the bulk modulus from various models. The calculation targets are the same 100 structures used in the structure optimization and molecular dynamics tasks. The bulk modulus is obtained by fitting the Birch-Murnaghan equation of state calculated by applying multiple strains to the input structure. To exclude unstable structures, any structure where the minimum volume obtained from the fitting deviated by more than 1% from the optimized volume was excluded. Therefore, the number of successful calculations varies by model, and this number is shown in parentheses above the bar graph for each model. As the figure shows, PFP cleared this criterion for 98 out of 100 structures, the same number as uma-1p1-odac. In terms of accuracy, it showed the second-best performance after eSEN-OAM. This suggests that PFP is an excellent model in terms of both calculation stability and accuracy.
![Figure 4. Evaluation results for the bulk modulus task. Left: Violin plot of the difference between predicted and correct values(ΔK = K – KDFT [3, 4]. Right: Comparison of MAE by model. The MAE and the number of successfully calculated structures out of 100 (in parentheses) are listed above each bar.](https://matlantis.com/wp-content/uploads/2025/10/6.png)
Figure 4. Evaluation results for the bulk modulus task. Left: Violin plot of the difference between predicted and correct values(ΔK = K – KDFT [3, 4]. Right: Comparison of MAE by model. The MAE and the number of successfully calculated structures out of 100 (in parentheses) are listed above each bar.
(5) Heat Capacity
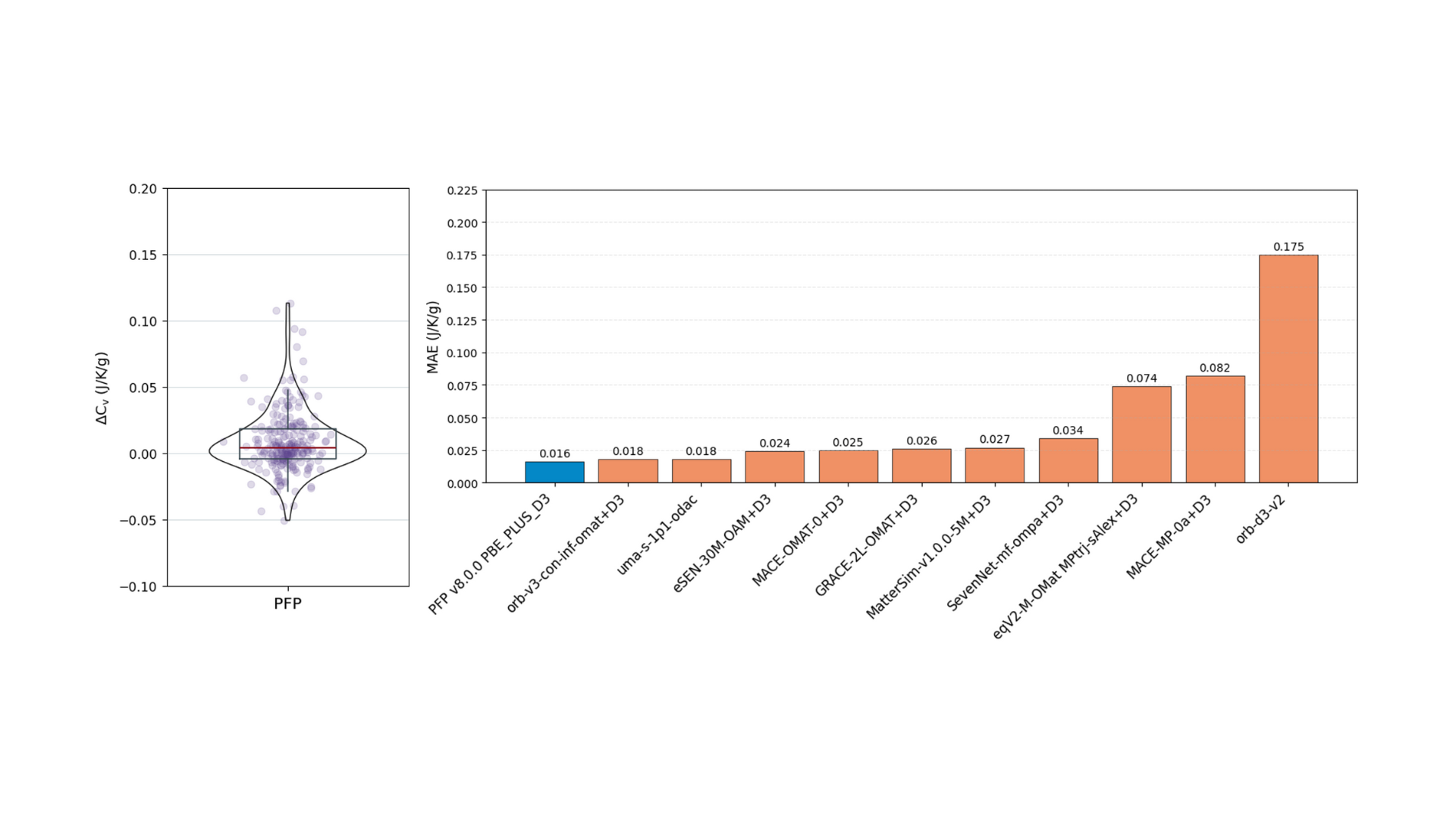
The specific heat calculation results are shown in Figure 5. The targets are 231 structures collected from the CoRE-MOF database. For these, the specific heat at 300K is evaluated after structure optimization, force constant calculation, and phonon calculation. PFP demonstrated excellent calculation accuracy for specific heat, with orb-v3-omat+D3 and uma-1p1-odac showing comparable accuracy.
Figure 5. Evaluation results for the specific heat task. Left: Violin plot of the difference between predicted and correct values (ΔCv= Cv– CvDFT). Right: Comparison of MAE by model. The MAE is listed above each bar.
(6) Host-Guest Interaction
The evaluation of host-guest interactions utilizes test data from the GoldDAC database to assess the energy and forces of CO 2/H2O interaction with 26 different MOFs [6]. The energy is calculated by subtracting the energies of the isolated MOF and gas molecule from the total energy of the adsorbed system. The forces are evaluated by comparing the forces on the entire MOF structure with the adsorbed gas molecule to the DFT reference values. Figures 6 and 7 show the evaluation results for interaction energy and forces by model, respectively. R, E, and W correspond to Repulsion, Equilibrium, and Weak-attraction, representing interactions at different reaction coordinates. 'all' represents the evaluation across all data points.
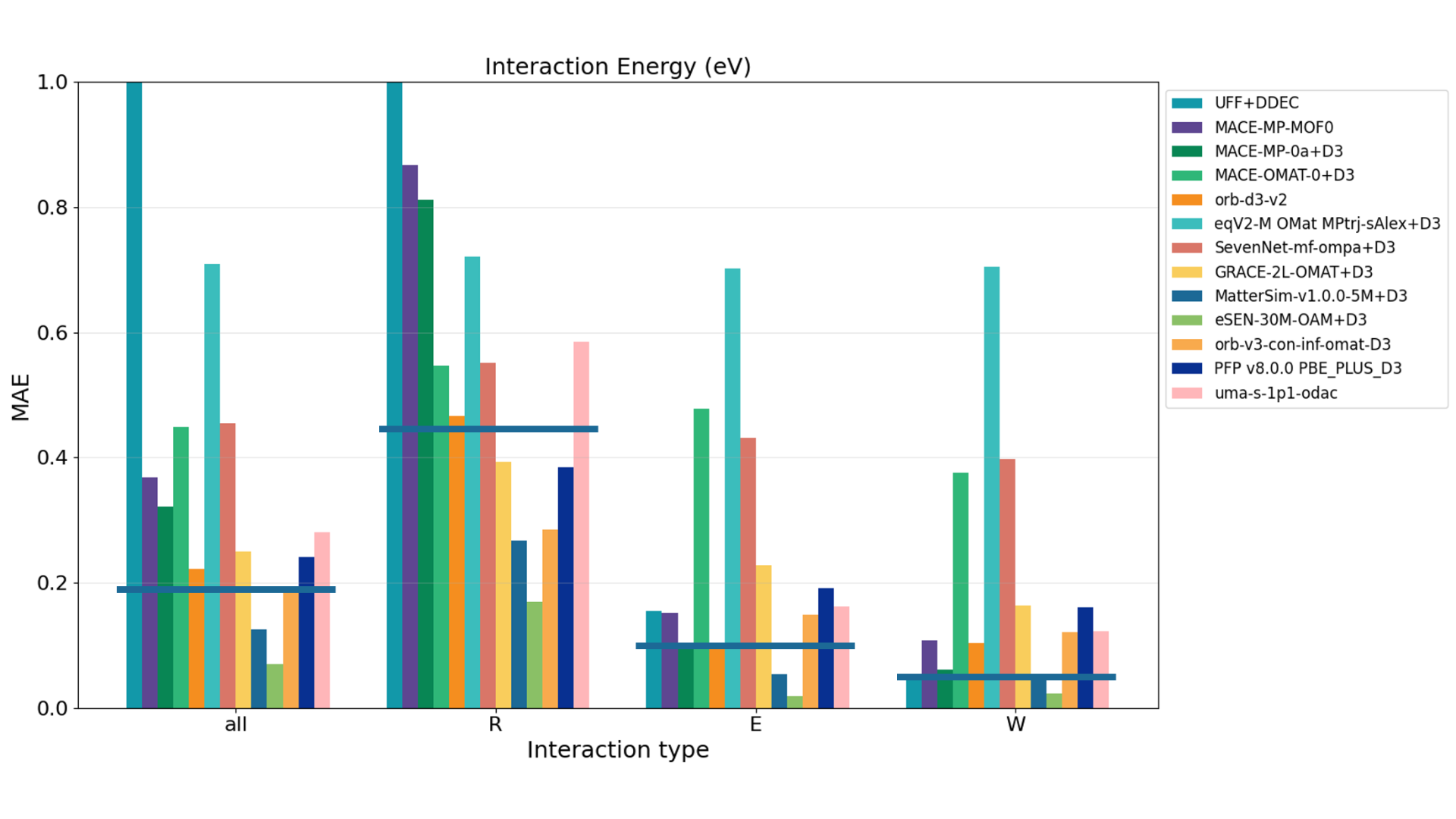
Figure 6. Evaluation results of interaction energy by model. The horizontal line in 'all' and for each reaction coordinate R, E, W indicates the result of MACE-DAC-1+D3, which is MACE-MP-0 fine-tuned on the GoldDAC dataset.
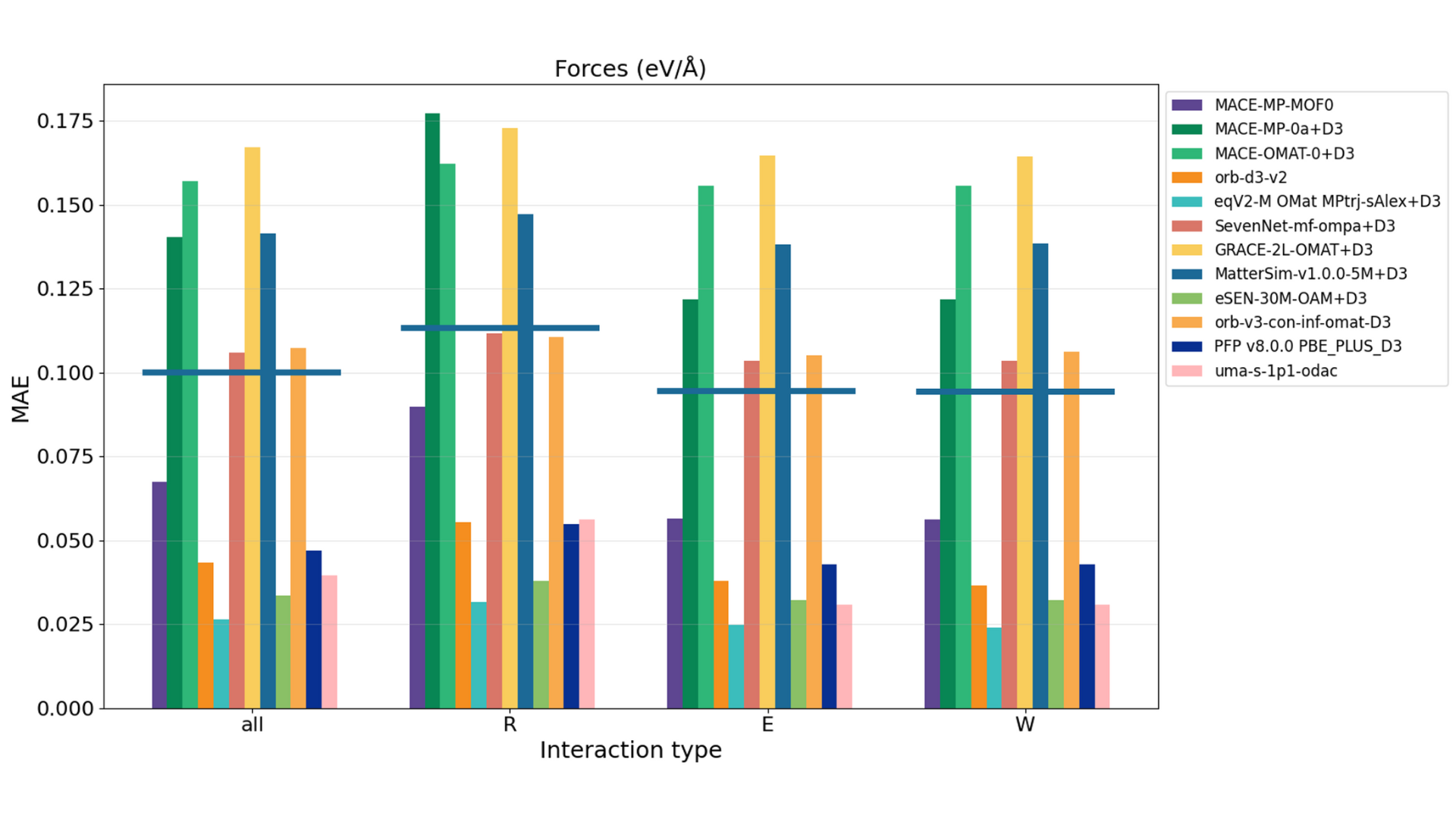
Figure 7. Evaluation results of forces by model. The horizontal line in 'all' and for each reaction coordinate R, E, W indicates the result of MACE-DAC-1+D3, which is MACE-MP-0 fine-tuned on the GoldDAC dataset.
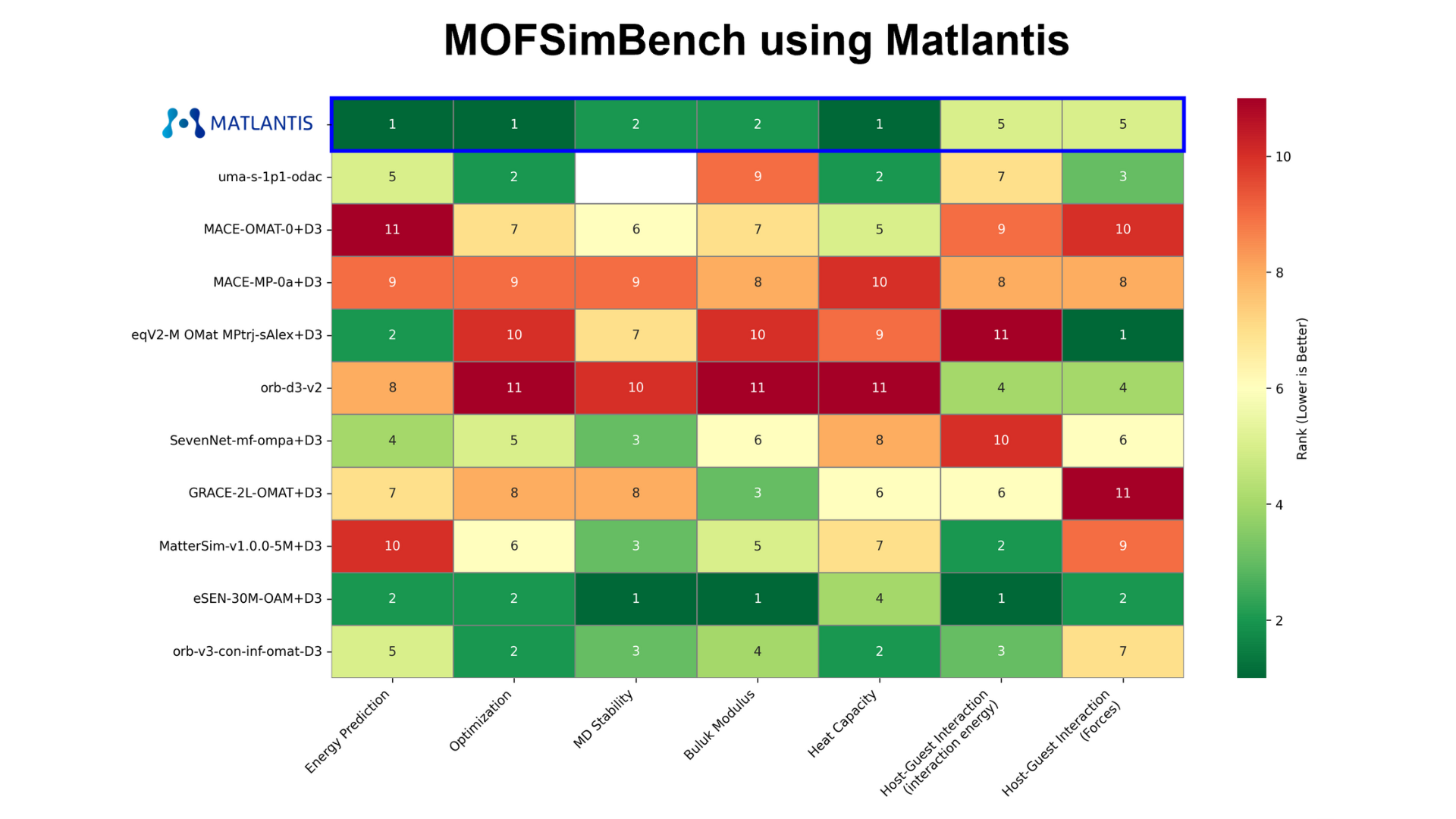
Figure 8 summarizes the ranked results from 1st to 11th place for each benchmark. Better rankings are colored in darker green. For the host-guest interaction task, the ranking for the 'all' reaction coordinate results is listed. While some models show excellent accuracy only for specific tasks, PFP and eSEN-OAM demonstrated consistently superior performance across all tasks. Following them, orb-v3-omat and uma-s-1p1 provided good results. On the other hand, the accuracy of non-conservative models that directly predict forces, such as orb-d3-v2 and eqV2-OMsA, was relatively low.
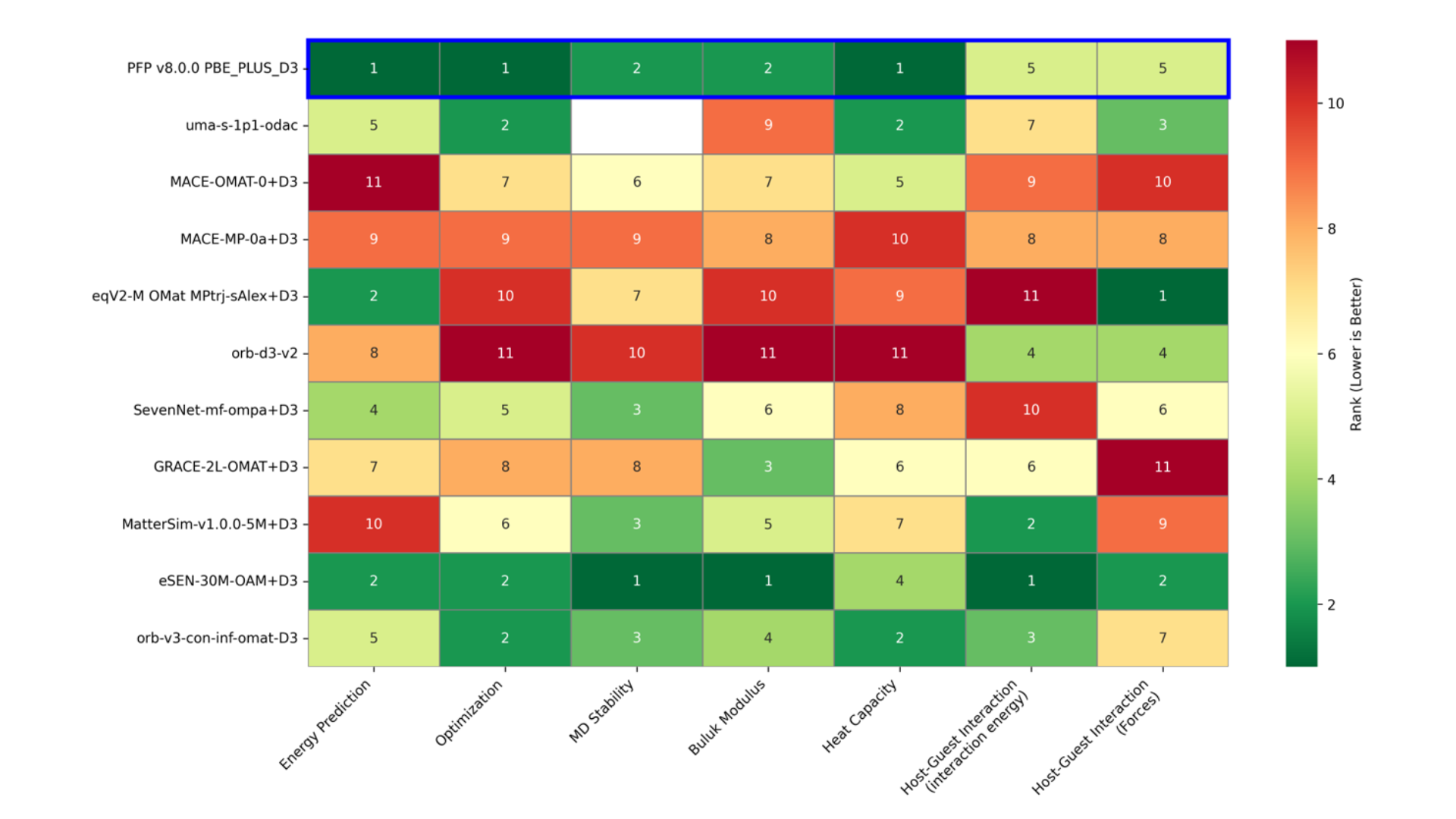
Figure 8. Ranking of each model in each task. The entry for uma-s-1p1 in the Molecular Dynamics Stability task is blank as it was not tested.
(7) Calculation Speed
When evaluating the practical performance of MLIPs, not only calculation accuracy but also computational speed is important. Let's compare the calculation speeds by combining the results reported in reference [1] with PFP's original speed benchmark. Figure 9 shows the calculation speeds of the major MLIPs reported in reference [1], where the inference time per step was evaluated using a high-performance environment with an NVIDIA H100 GPU. As shown in the figure, eSEN-OAM, which demonstrated consistently high accuracy across multiple tasks, is slower than other models, with a speed of about 280 ms per step. This slower speed is due to the model’s large size of approximately 30 million parameters. Compared to the mid-sized model MatterSim-v1-5M (about 4.5 million parameters), it takes roughly 3.25 times longer.
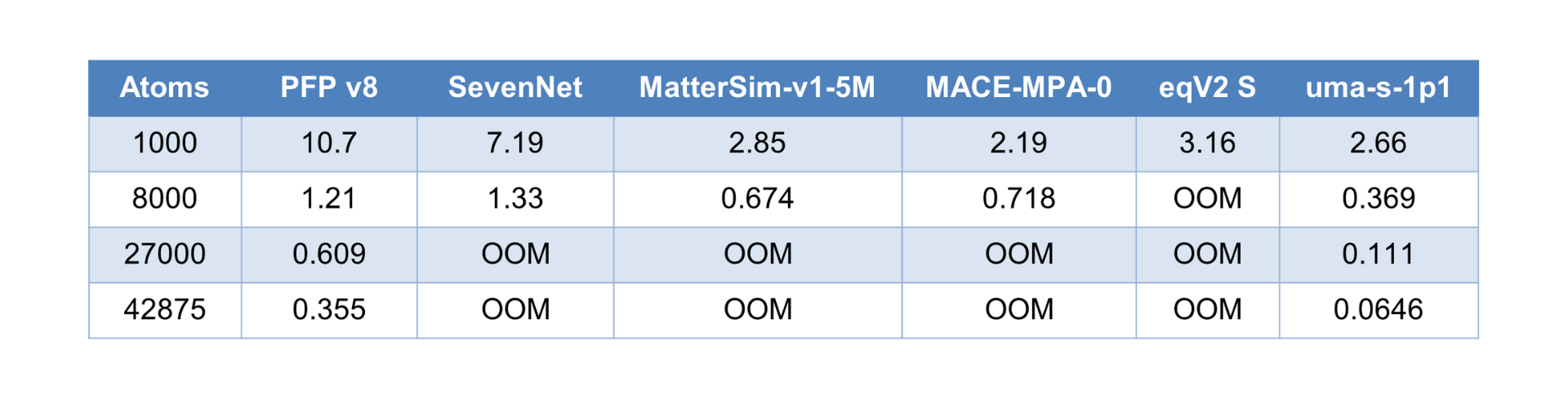
Although the calculation conditions and environments differ, Table 1 shows the number of inferences per second for various input structure sizes for PFP on Matlantis and other MLIPs measured in an A100 GPU environment. Dispersion correction is not considered here. For a structure with 1000 atoms, PFP can perform inference about 3.75 times faster than MatterSim-v1-5M. Therefore, it can be inferred that PFP is significantly faster than eSEN-OAM. We also confirmed that PFP is about 4 times faster than uma-s-1p1 on an A100 GPU environment for a 1000-atom structure.
Furthermore, while reference [1] applies torch-dftd to all models for dispersion correction, Matlantis accelerates this computation as well by utilizing the PFVM inference engine. As a result, when dispersion correction is required, Matlantis can perform calculations more efficiently.
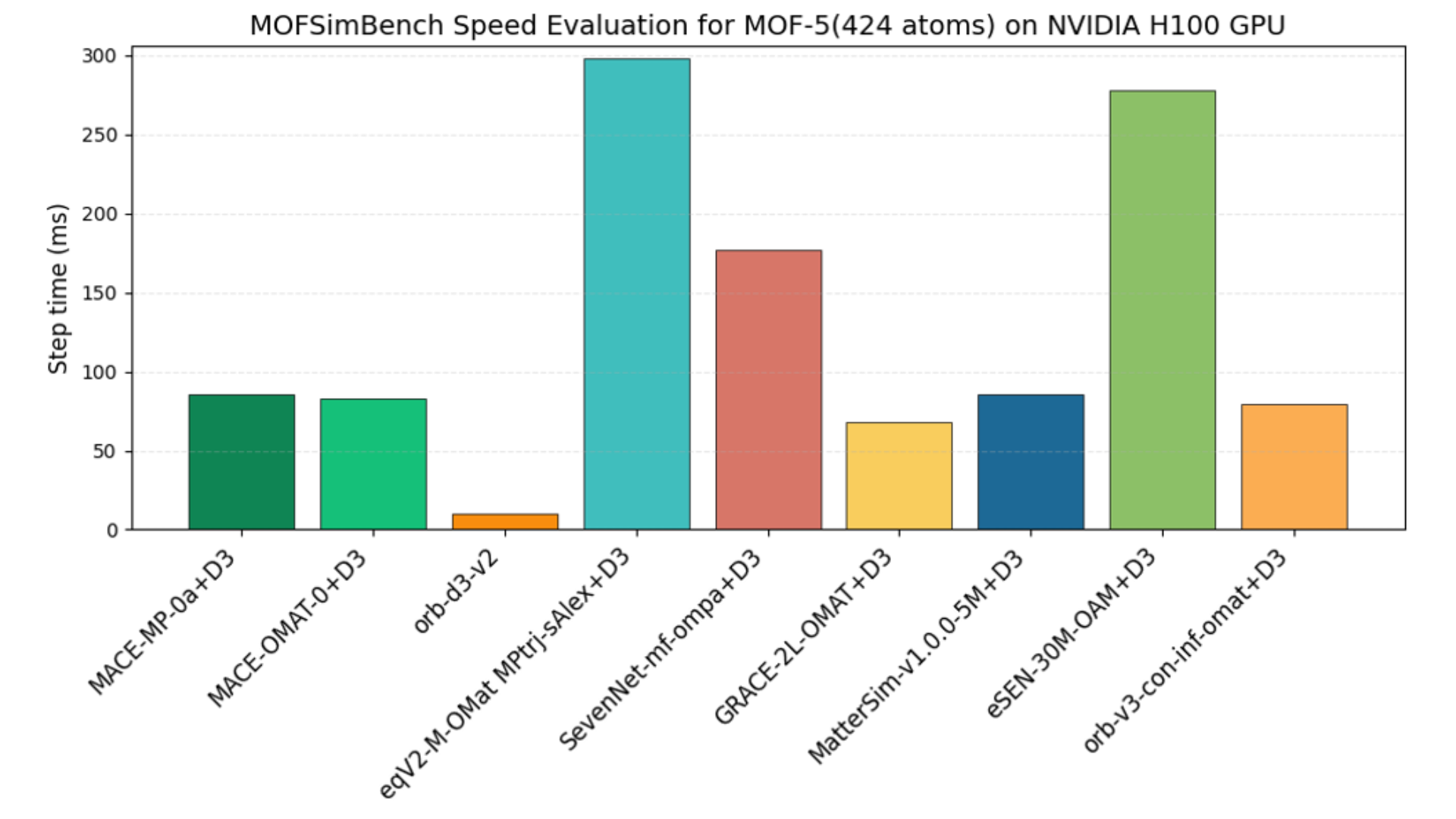
Figure 9. Comparison of calculation speeds of major general-purpose MLIPs reported in reference [1]. A 1000-step structure optimization was performed on the MOF-5 structure (424 atoms), and the average inference time per step was calculated.
Table 1. Number of inferences per second for different input structure sizes. An A100 GPU was used for the evaluation of open-source MLIPs. OOM (Out Of Memory) corresponds to cases where calculations for larger structure sizes could not be executed due to memory errors. Dispersion correction is not considered.
Summary
In this article, we evaluated the performance of PFP on the MOF material MLIP benchmark, MOFSimBench, and compared the results with the accuracy and speed benchmarks of other MLIPs reported in reference [1]. As a result, we confirmed that PFP v8.0.0 PBE PLUS D3, implemented in Matlantis, and eSEN-30M-OAM (with dispersion correction using torch-dftd), provided by Meta, both exhibited consistently excellent accuracy across all tasks. In particular, PFP showed top performance in energy prediction, structure optimization stability, and specific heat evaluation. Focusing on calculation speed, the results from reference [1] and our own speed benchmarks suggest that PFP is significantly faster than eSEN-OAM. Furthermore, when compared to the latest general-purpose MLIP, UMA (uma-s-1p1 task_name=odac), we demonstrate that PFP is superior in both accuracy and speed within the scope of this verification.
Based on these findings, we conclude that PFP achieves both high accuracy and high-speed computation for exploring MOF materials, and that it is a highly practical model compared with existing MLIPs. As symbolized by this year's Nobel Prize in Chemistry, MOFs are extremely important materials for realizing a sustainable society. We will continue to advance Matlantis's technological development to accelerate MOF material development and further contribute to society.
References
[1] https://arxiv.org/abs/2507.11806
[2] https://github.com/pfnet-research/torch-dftd
[3] https://matlantis.com/ja/product/about-pfp/
[4] https://arxiv.org/abs/2506.23971
[5] https://github.com/Andrew-S-Rosen/QMOF?tab=readme-ov-file
[6] https://chemrxiv.org/engage/chemrxiv/article-details/6759b06df9980725cfbc8cef
-




 URL
URL
Copied
Latest Articles
NEW
Learning AI materials simulation to accelerate research at the Fukui Kenichi Memorial Research Center, Kyoto University - Working with ENEOS to design the "best CO2 adsorbent"

NEW
Explainer : Why Did the AI Predict That ? Uncovering Atomic-Level Interpretability through PFP Descriptors and Shapley Values

Materials Informatics Explainer computational chemistry

Writing SMILES from scratch
Explainer computational chemistry

Nagoya University × Matlantis Case Study:“Advanced Experiments for Frontier Technologies and Sciences” —A Four-Day Intensive Course That Sparked Experimental Students’ Curiosity Through AI Simulation
Interview computational chemistry

Introduction to Machine Learning Interatomic Potentials (MLIPs): A Game Changer in Materials Simulation

Machine Learning Interatomic Potentials Explainer