
Matlantisで計算可能な材料は多岐に渡りますが、その中でも金属有機複合体(Metal-Organic Frameworks:MOF)は触媒、CO2貯蔵など幅広い用途をもつ重要な材料です。1990年代に見出されたこの材料は既に産業化も進められており、持続可能な社会を持続するために不可欠な材料として世界的にも注目されています。実際、2025年のノーベル化学賞はこの豊かな化学的鉱脈を見出した3人の研究者に授与されました。本稿では、MatlantisのMOF材料に対する予測性能を検証するため、2025年7月に公開されたMOF材料のベンチマークMOFSimBenchに対して計算を行った結果を報告します。また、他のオープンソースの機械学習ポテンシャルとの比較結果も記載しています。
MOFSimBenchの概要と計算条件
MOFSimBenchはMOFに関する既存のデータベースであるQMOF, MOSAEC-DB, IZA, CURATED-COFから多様性を重視して収集した100の構造群と、CoRE MOF, GoldDACデータベースの構造群を用いて計算を行っています[1]。ベンチマークとして設定されているのは構造最適化、分子動力学計算、体積弾性率、比熱、ホスト-ゲスト相互作用の5種のタスクです。これらに対し、各種オープンソースの機械学習ポテンシャル(MLIP)の性能を評価しています。また、補足的にQMOFデータベースのエネルギー予測精度とモデルごとの計算速度も検証されています。正解データとなる計算はPBE汎関数を用いたDFTで行われています。比較検討されているMLIPは、MACE、Orb、SevenNet、 MatterSim、 eSEN-OAMなど著名なモデルが含まれています。これらのモデルの予測結果に分散力補正を考慮するため、Preferred Networks社がオープンソースで公開しているtorch-dftdパッケージが用いられています[2]。
本稿では、MOFSimBenchの検証を実施しました。また、文献[1]の結果と弊社が既に取得していたベンチマークを元に、計算速度に関する議論も行いました。使用したモデルは、Matlantisが提供する2025年10月現在の最新モデルPFP v8.0.0に分散力補正を考慮したPBE PLUS D3モードです。PFPは、独自の推論エンジンPFVMによって高速化されています[3]。さらに、MOFSimBenchでは検証されていないMeta社が提供するUMA (Universal Models for Atoms)の小型モデルについても検証を行いました(モデル名:uma-s-1p1)[4]。UMAは計算したい材料の種類に応じてタスクと呼ばれるパラメータを切り替える必要があり、ここではodacをタスクとして設定しました。uma-s-1p1の計算にはNVIDIA Tesla T4 GPUを用いました。それ以外のモデルの予測精度は、参考文献に記載されている数値を引用しました。
(1) エネルギー予測
図1に、QMOFデータベース[5]に格納されている約2万件の構造に対するPFPの予測結果と各MLIPモデルのエネルギー予測性能の比較を示します。左図のパリティプロットから明らかなように、PFPはDFTの結果と優れた一致を示しており平均絶対値誤差(Mean Absolute Error, MAE)は0.006 eV/atomでした。他のモデルと比べると、eSEN-OAMやequiformerV2といったモデルより優れた結果となっています。
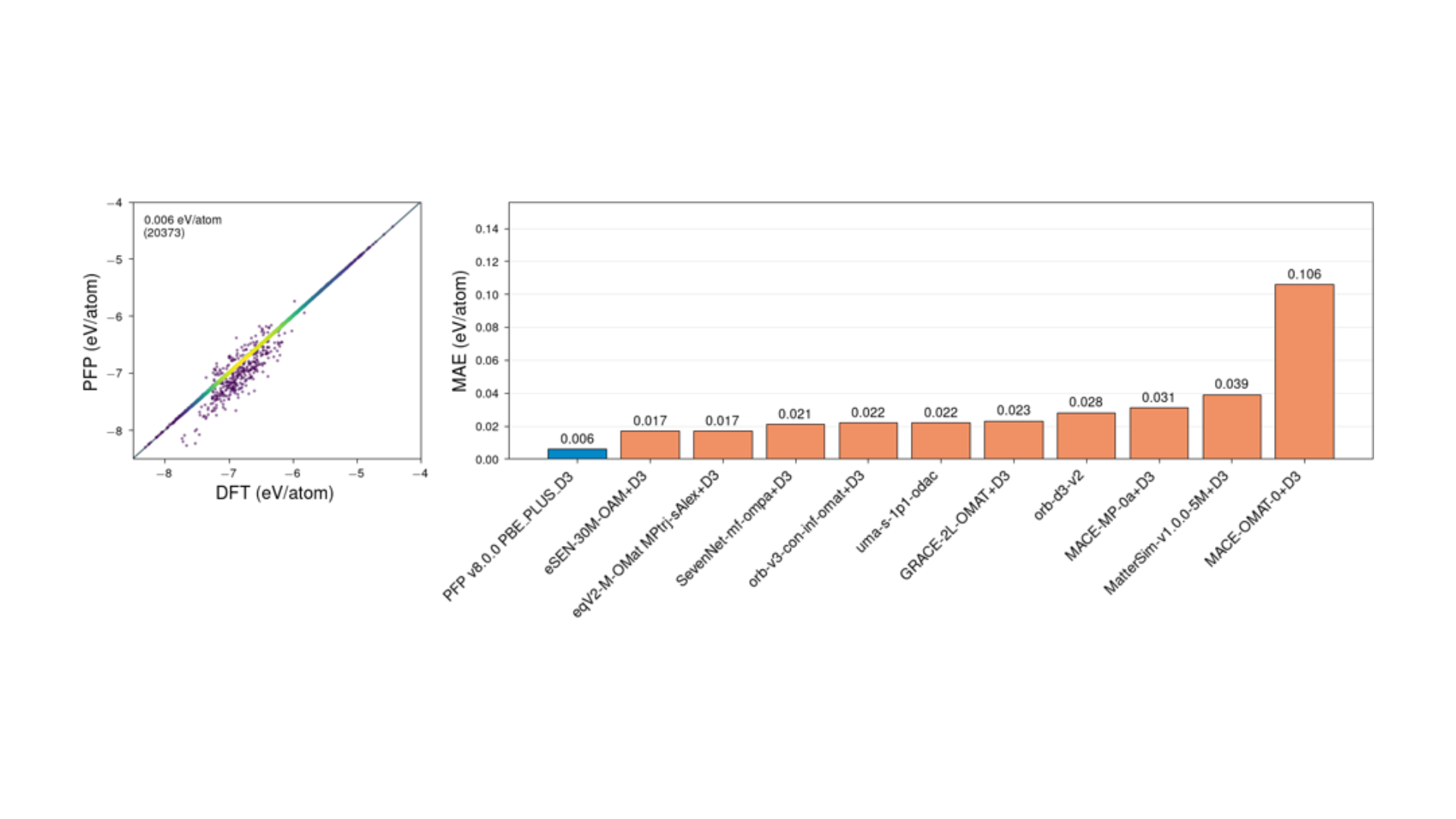
図1. QMOFに対するエネルギー予測精度の評価結果。左図:PFPとDFTのパリティプロット。右図:予測誤差(MAE)のモデルごとの比較。
(2) 構造最適化
構造最適化タスクでは、100件の構造(83個のMOF構造、7個のCOF構造、10個のゼオライト構造)に対してMLIPで構造最適化を行い、DFTで得られている最適化構造との体積変化率を評価します。図2の左側には100個の最適化構造のPFPとDFTの体積変化率をバイオリンプロットで示しています。グレーで塗られた部分は、±10%の領域に対応しています。ご覧の通り、PFPは100構造に対し92構造がこの領域に収まるという結果を得ました。図3の右側の棒グラフでは、各モデルで±10%の領域に収まった構造数を比較しています。この図から分かるように、PFPはこれらのMLIPの中でも最高の水準を示しています。次いで、orb-v3-omat+D3、eSEN-OAM、uma-s-1p1が優れた性能を示しました。
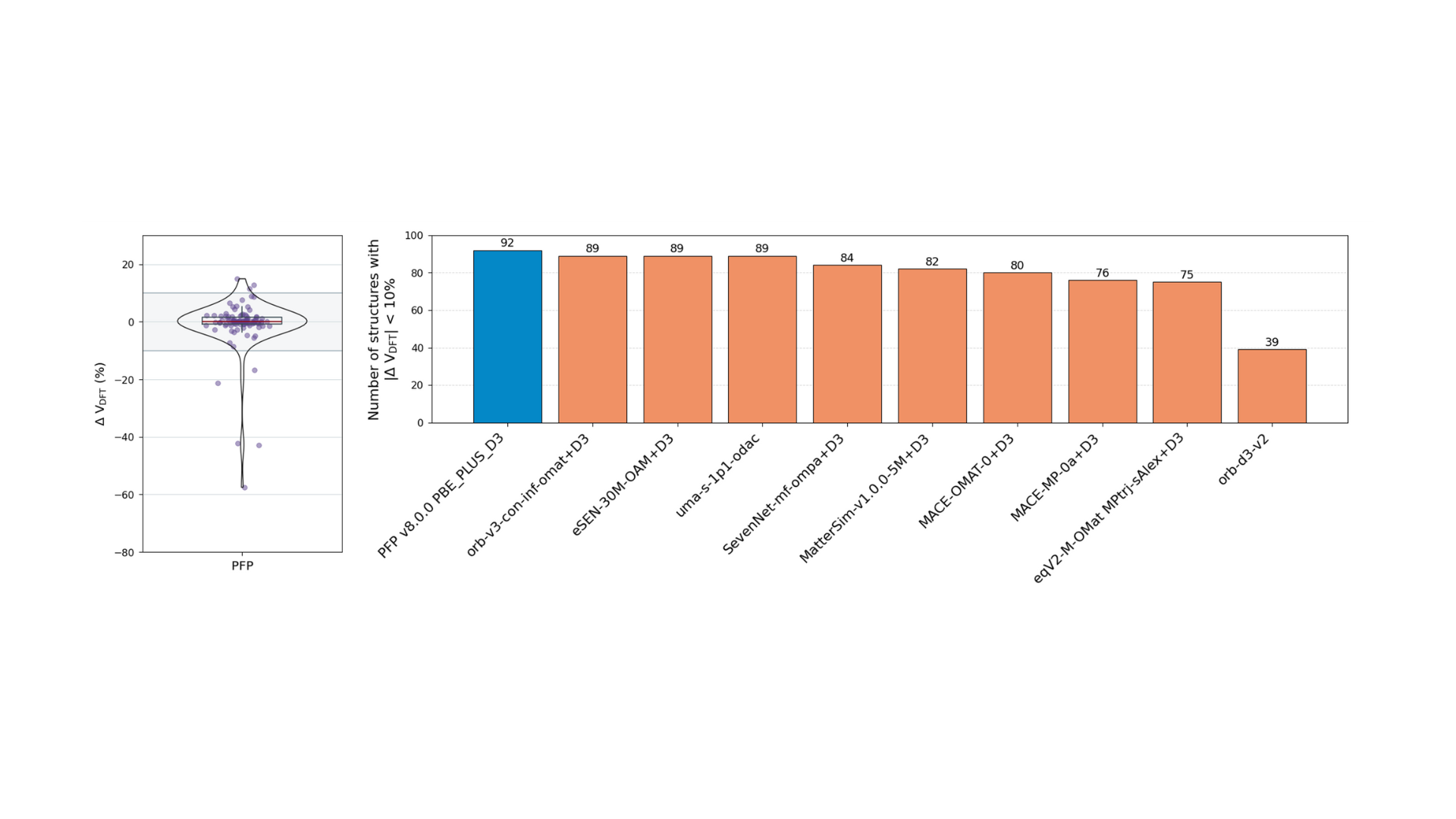
図2. 構造最適化タスクにおける評価結果。左図:体積変化率(ΔVDFT = 1 – V / VDFT )のバイオリンプロット。右図:|ΔVDFT| < 10%となる構造数の比較。
(3) 分子動力学計算
分子動力学計算による安定性も評価しました。構造最適化タスクと同様に、100個の構造に対して分子動力学計算が行われています。構造最適化とNVT計算によって平衡化された構造に対して、300K、1barの条件下で50psのNPTシミュレーションが行われ、その初期構造と最終構造の体積変化率を評価しています。構造最適化タスクと同様に、体積変化率の絶対値が10%未満の構造数をモデルごとに比較したのが、右側の棒グラフです。eSEN-OAM、 PFP、 orb-v3-omat+D3といったモデルが上位となっています。なお、Testa T4 GPUを用いた計算ではuma-s-1p1の十分な計算速度が得られず分子動力学計算タスクは評価していません。
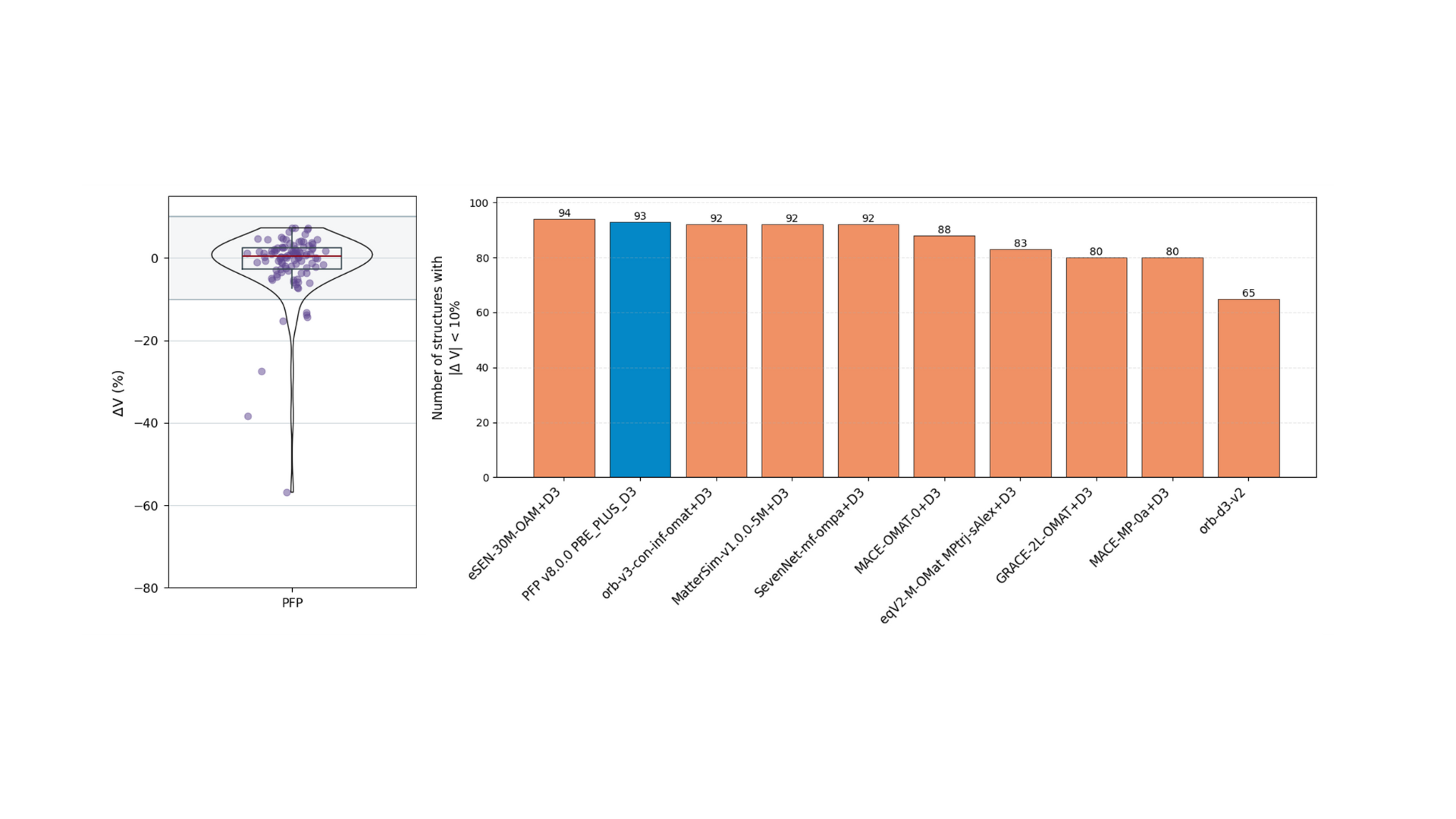
図3. 分子動力学計算タスクにおける評価結果。左図:体積変化率(ΔV = 1 – Vfin / Vini)のバイオリンプロット。右図:|ΔV| < 10%となる構造数の比較。
(4) 体積弾性率
MOFSimBenchでは、MOF材料のバルク物性の予測性能を体積弾性率および比熱で評価しています。図4に示すのは、体積弾性率の各種モデルの予測結果です。計算対象は構造最適化タスクと分子動力学計算タスクと同じ100構造です。体積弾性率は、入力構造に対して複数の歪を適用して計算したBirch-Murnaghan状態方程式のフィッティングから得ています。不安定な構造を除外するため、フィッティングで得られた最小体積が最適化後の体積から1%より外れた場合、その構造は除外しています。そのためモデルによって計算に成功した数が異なり、各モデルに対する棒グラフの上部に括弧書きで記載しています。図に示す通り、PFPは100構造中98構造に対してこの基準をクリアし、これはuma-1p1-odacと同数となっています。精度面ではeSEN-OAMに次ぐ性能を示しています。PFPが計算の安定性、精度ともに優れたモデルであることが示唆されます。
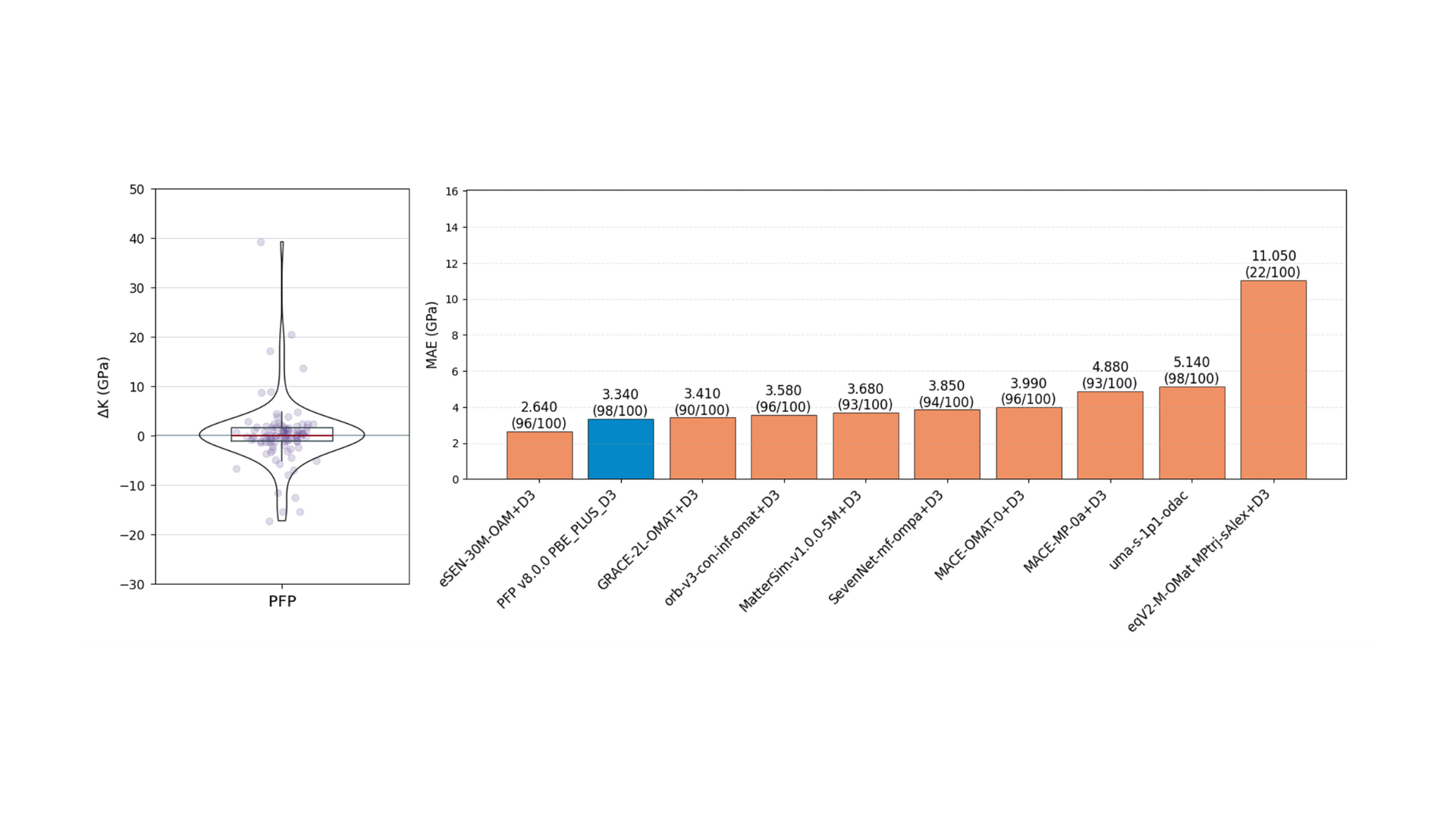
図4. 体積弾性率タスクにおける評価結果。左図:予測値と正解値の差分(ΔK = K – KDFT)のバイオリンプロット。右図:MAEのモデルごとの比較。各棒の上部には、MAEおよび括弧書きで100構造に対して計算に成功した構造数を記載。
(5) Heat Capacity
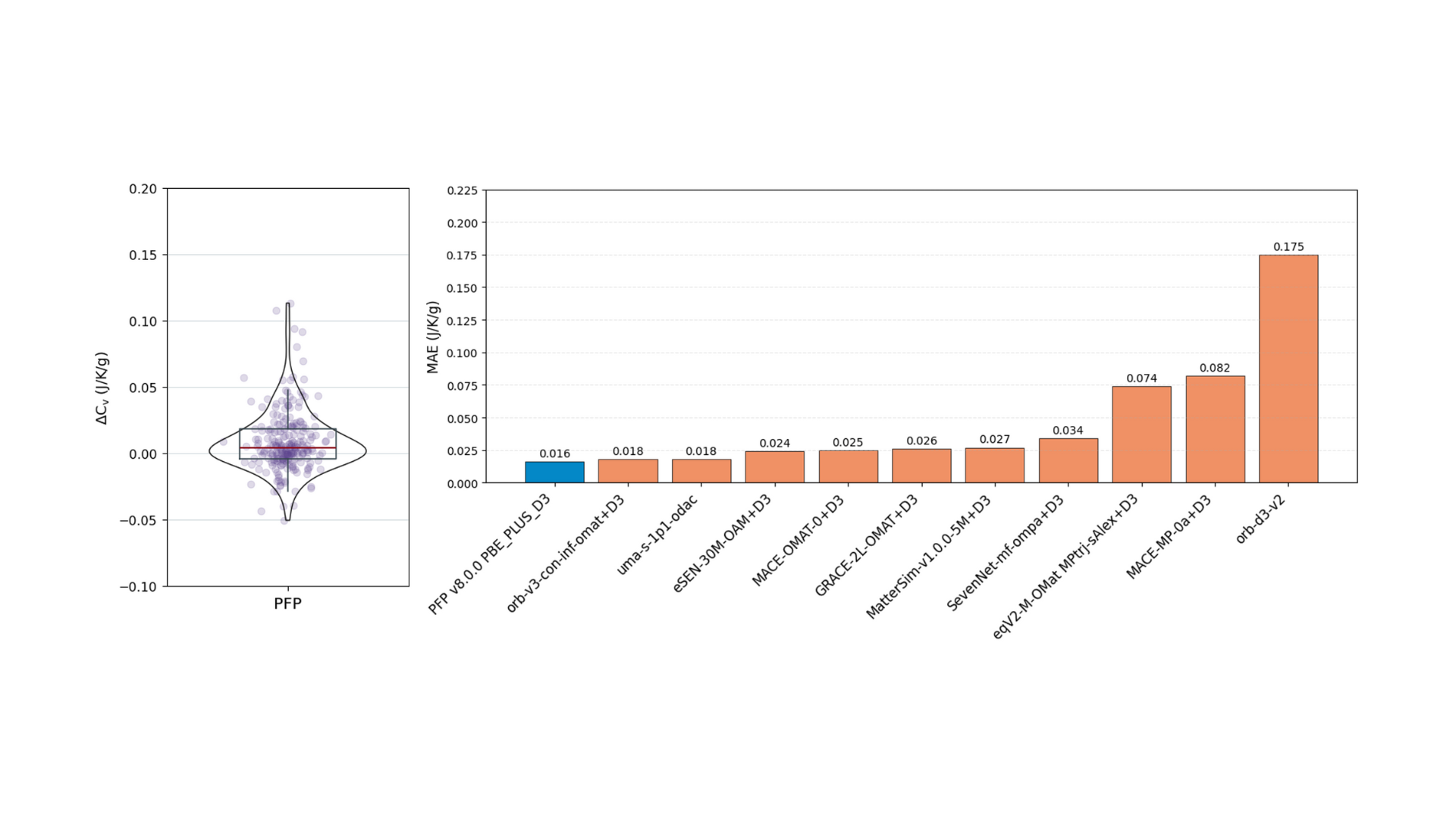
比熱の計算結果を図5に示しました。計算対象は、CoRE-MOFデータベースから収集されている231件の構造です。これらに対し、構造最適化、力定数の算出、フォノン計算を行い300Kにおける比熱を評価しています。比熱計算に対してPFPは優れた計算精度を示しており、orb-v3-omat+D3、uma-1p1-odacも同等の精度でした。
図5. 比熱タスクにおける評価結果。左図:予測値と正解値の差分(ΔCv = Cv – CvDFT)のバイオリンプロット。右図:MAEのモデルごとの比較。各棒の上部にはMAEを記載。
(6) ホスト-ゲスト相互作用
ホスト-ゲスト相互作用の評価にはGoldDACデータベースのテストデータを活用し、26種のMOFに対するCO2/H2O相互作用のエネルギーと力を評価しています[6]。エネルギーは、吸着状態の全系のエネルギーからMOF単体、ガス分子単体のエネルギーを差し引き算出しています。力は、ガス分子が吸着したMOF構造全体の力をDFTの参照値と比較しています。図6、7はそれぞれ相互作用エネルギーと力のモデルごとの評価結果であり、R、E、WはそれぞれRepulsion、Equilibrium、Weak-attractionに対応し、異なる反応座標での相互作用を意味しています。allはそれらすべてを含む全データポイントでの評価結果です。 結果を見ると、eSEN-OAM、MatterSimのMAEがどの指標でも低くなっています。また、指標によって多少のばらつきはあるものの、PFPとuma-1p1-odacは概ね同程度の性能を示しているようです。なお、本計算は入力構造に対してそのままエネルギーや力を計算しており、MLIPによる構造最適化は行われていません。
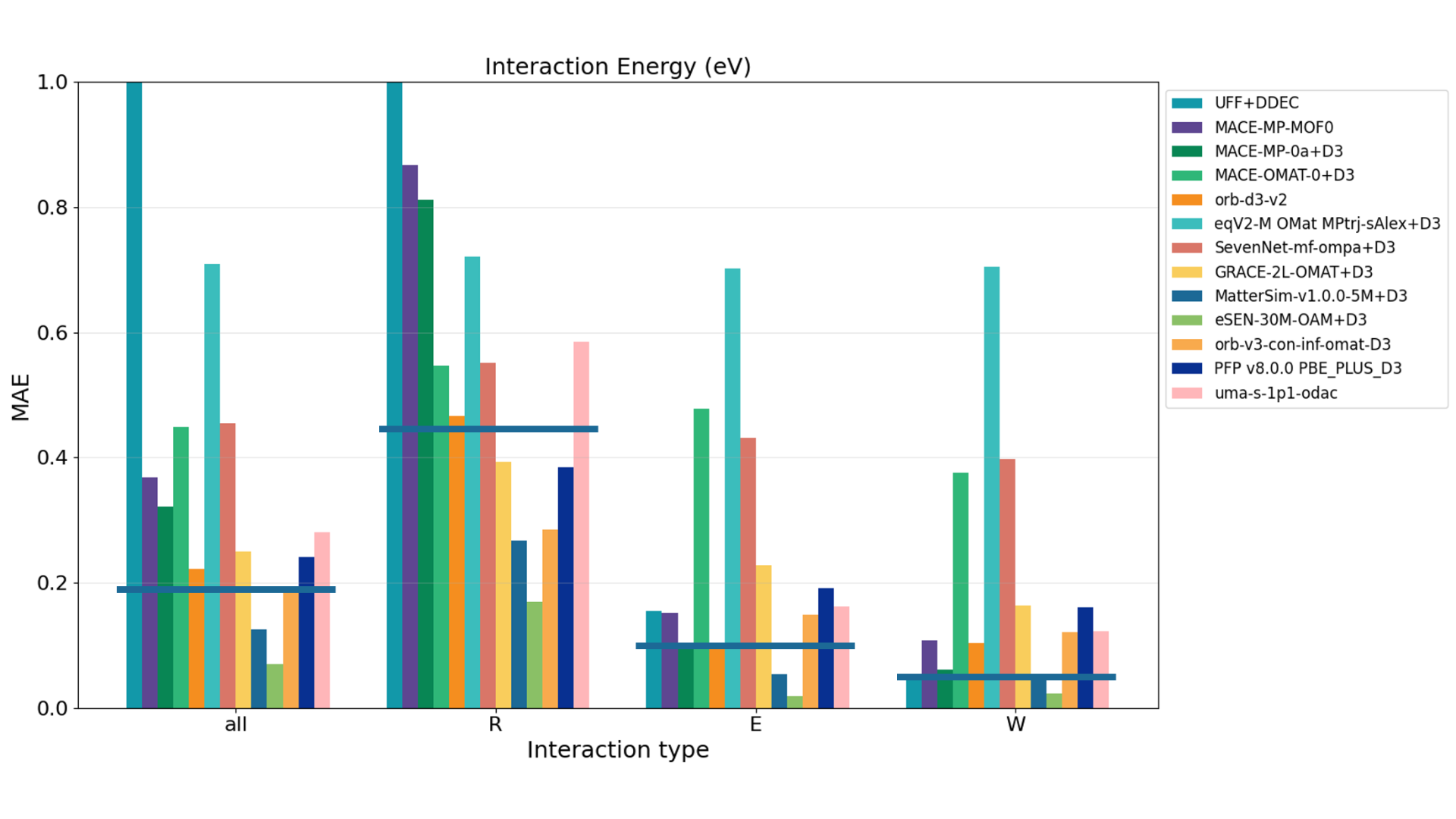
図6. モデルごとの相互作用エネルギーの評価結果。allと各反応座標R、E、Wにおいて横に引かれている線は、GoldDACデータセットでMACE-MP-0をファインチューニングしたMACE-DAC-1+D3の結果を示す。
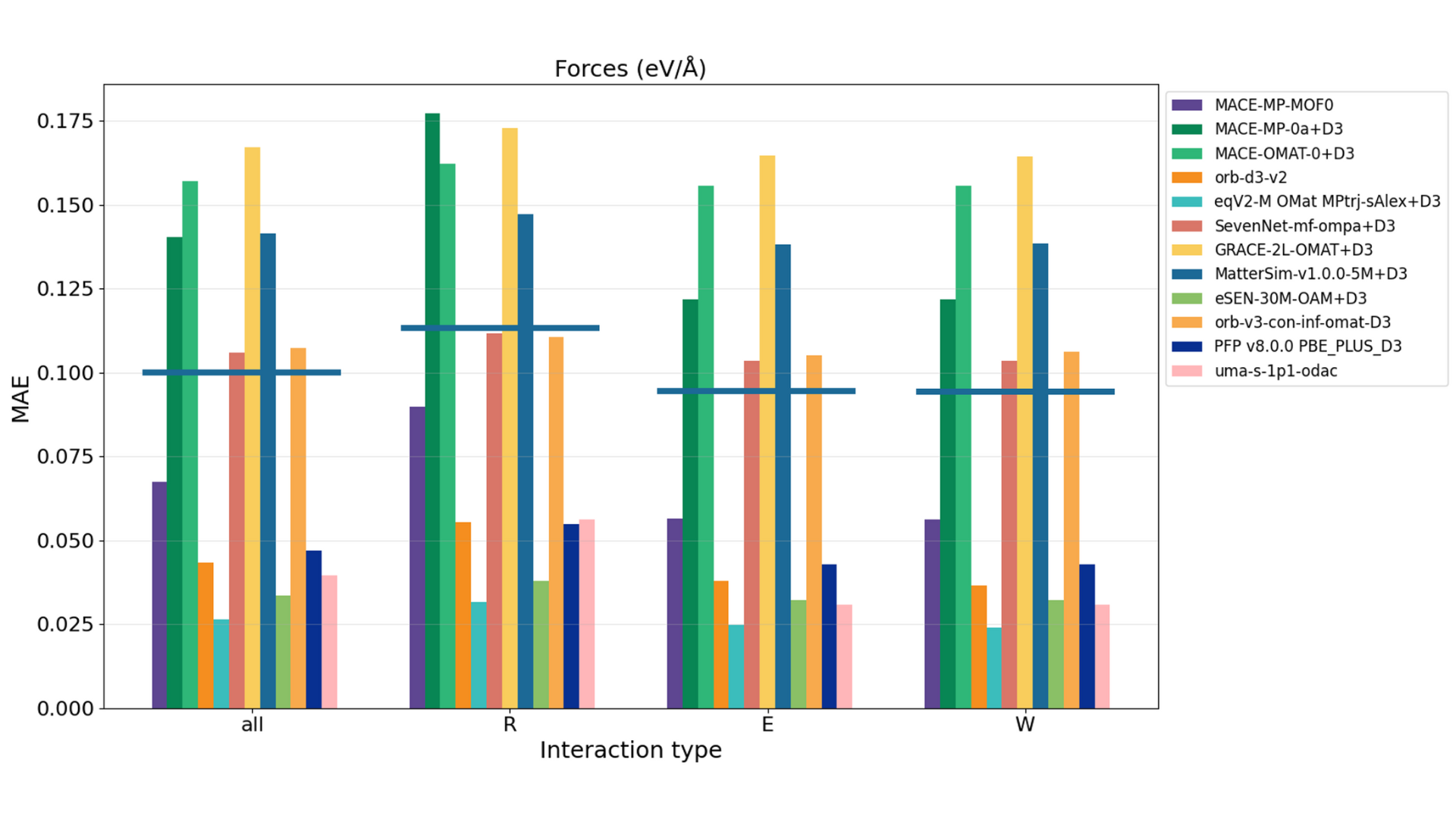
図7. モデルごとの力の評価結果。allと各反応座標R, E, Wにおいて横に引かれている線は、GoldDACデータセットでMACE-MP-0をファインチューンしたMACE-DAC-1+D3の結果を示す。
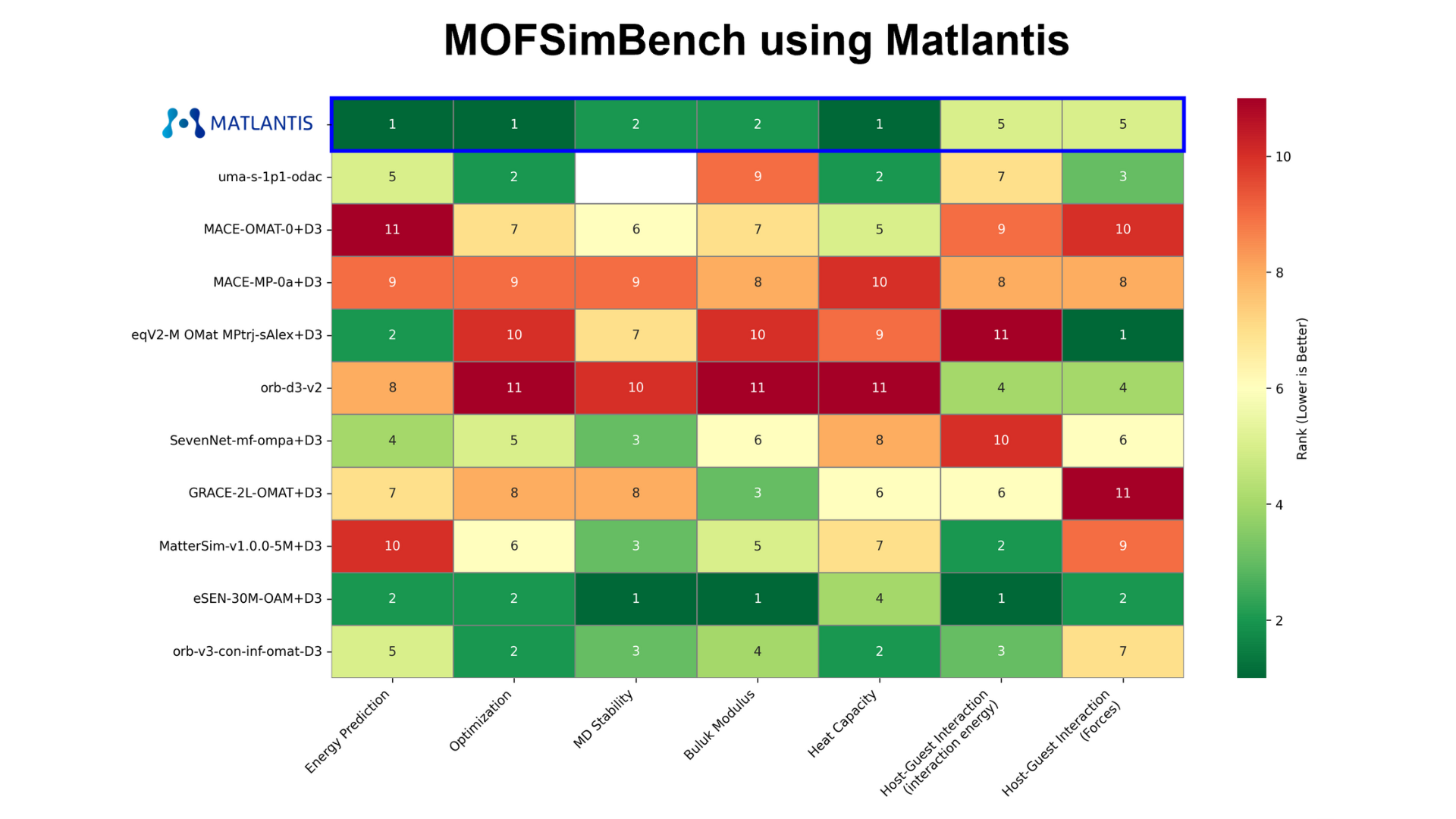
各ベンチマークの1位から11位まで順位付けされた結果を図8にまとめました。良い順位ほど濃い緑色で色付けされています。ホスト-ゲスト相互作用タスクでは、反応座標がallの結果の順位を記載しています。特定のタスクにのみ優れた精度を示すモデルが散見されますが、PFP、eSEN-OAMがすべてのタスクに置いて一貫して優れた性能を示しました。次いでorb-v3-omat、uma-s-1p1が良い結果を与えました。一方、力を直接予測するnon-conservativeモデルであるorb-d3-v2、eqV2-OMsAの精度は比較的低くなっています。
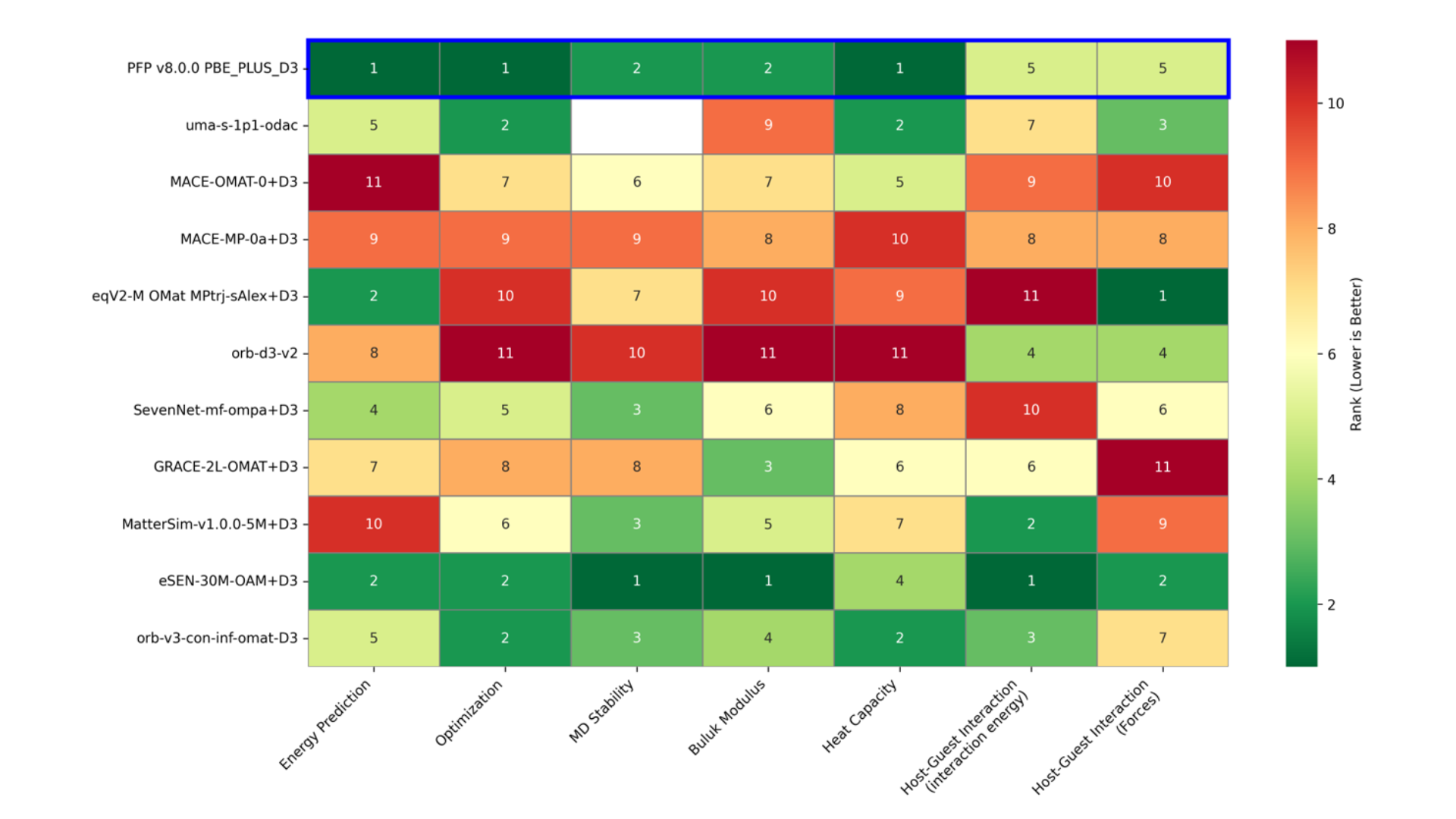
図8. 各タスクにおける各モデルの順位。分子動力学計算タスク(MD Stability)のuma-s-1p1は未検証のため空欄としている。
(7) 計算速度
MLIPの実用的な性能を評価する上では、計算精度に加え計算速度のベンチマークも重要です。文献[1]で報告されている速度ベンチマークと、社内で独自に取得したPFPの速度ベンチマークを組み合わせ、計算速度を比較してみましょう。図9は文献[1]で報告されている主要なMLIPの計算速度を示しており、NVIDIA H100 GPUという優れた計算環境を用いて1 stepあたりの推論時間を評価しています。図から明らかなように、複数タスクで一貫して高精度を示したeSEN-OAMの計算速度は1stepあたり約280msと他のモデルと比べ遅くなっています。これは本モデルが約3000万パラメータを持つ大型のMLIPであることに起因しています。約450万パラメータのMatterSim-v1-5Mと比較すると約3.25倍の時間を要しています。
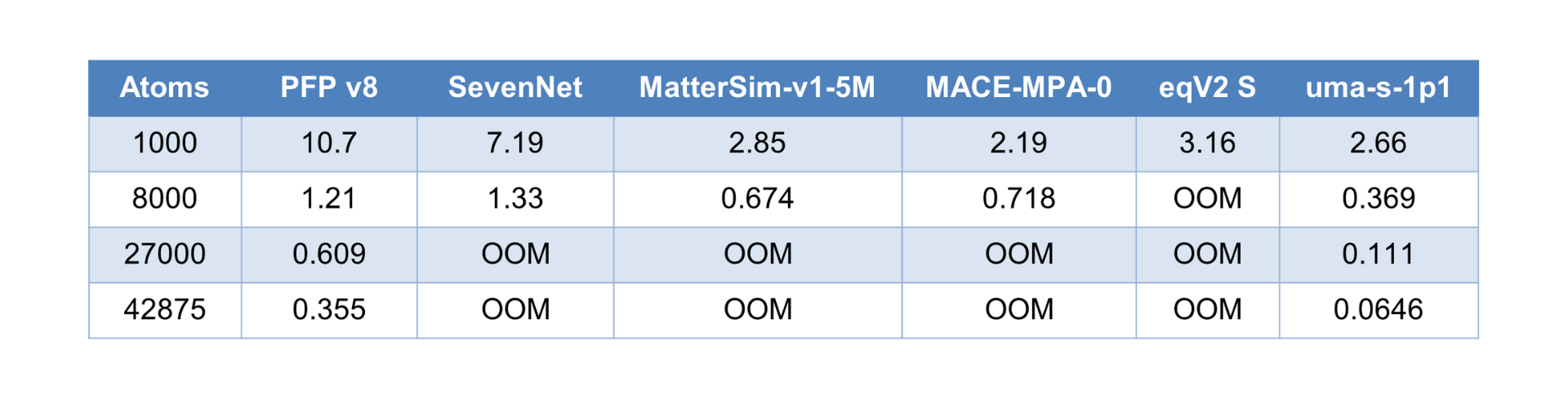
異なる計算条件・計算環境にはなりますが、Matlantis上のPFPおよびNVIDIA A100 GPU環境で測定した他のMLIPの入力構造サイズごとの1秒あたりの推論回数を表1に示します。入力サイズが1000原子の構造の場合、MatterSim-v1-5Mと比較するとPFPは約3.75倍高速に推論が可能です。そのため、eSEN-OAMと比較するとPFPが大幅に高速であることが推測されます。また、A100 GPU環境でのuma-s-1p1と比較すると、1000原子構造に対してPFPが約4倍高速であることも確認できました。
さらに、文献[1]では分散力補正にtorch-dftdを全モデルに対し適用していますが、Matlantisはこの計算もPFVMを用いて高速化しています。そのため分散力補正を要する場合には、Matlantisはより効率的に計算を行うことができます。
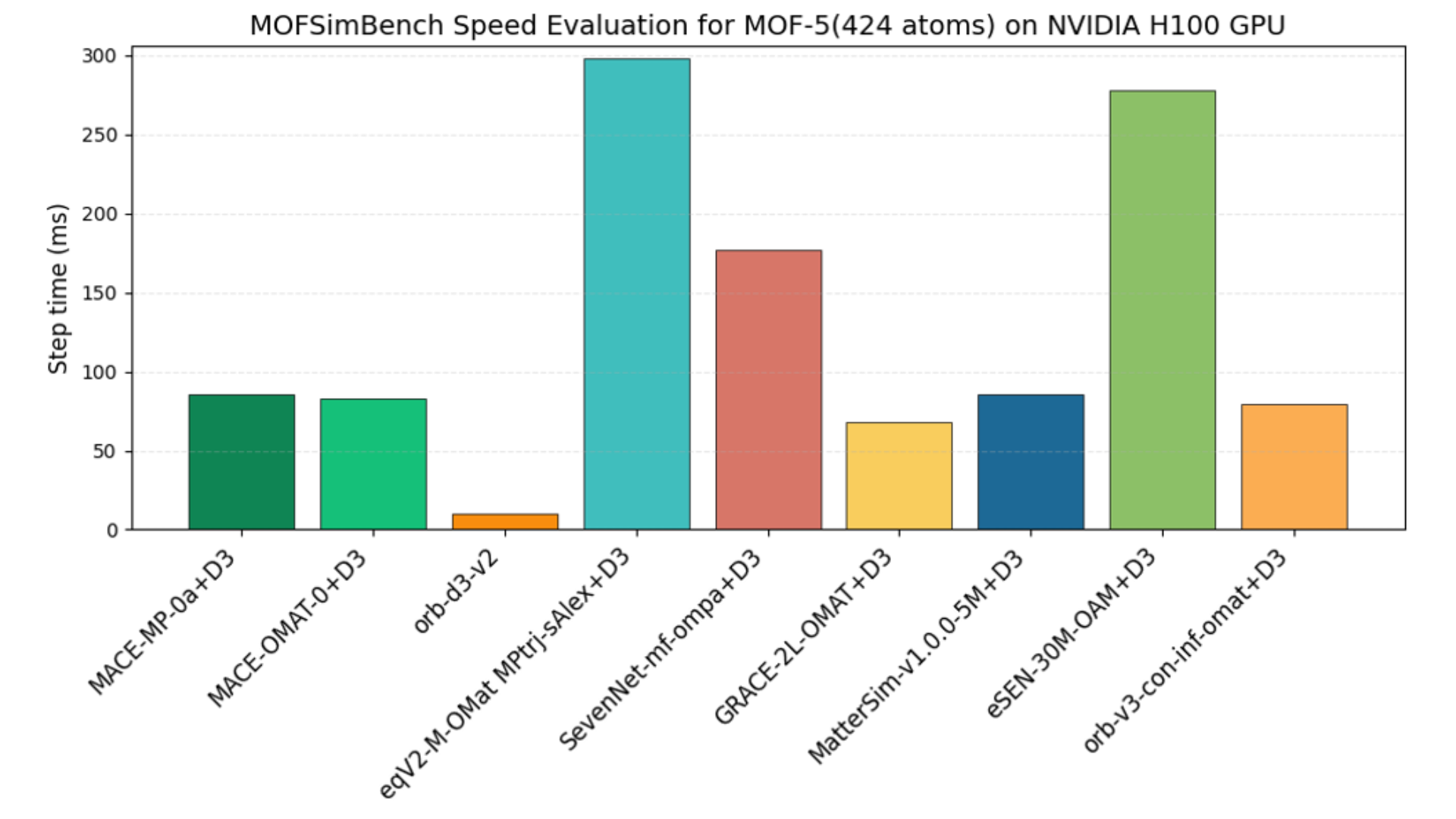
図9. 文献[1]で報告されている主要な汎用MLIPの計算速度比較。MOF-5 (424原子) を対象として1000 stepの構造最適化を行い、1 stepあたりの平均的な推論速度を算出。
表1. 入力構造サイズに対する1秒あたりの推論回数。オープンソースのMLIPの評価にはA100 GPUを利用。OOM (Out Of Memory)はメモリーエラーでこれ以上の構造サイズの計算が実行できないことに対応。分散力補正は考慮していない。
まとめ
本稿では、MOF材料のMLIPベンチマークであるMOFSimBenchに対してPFPの性能検証を行い、文献[1]で報告されているMLIPの精度・速度検証結果と比較しました。結果としてMatlantisに実装されているPFP v8.0.0 PBE PLUS D3と、Meta社が提供するeSEN-30M-OAM (torch-dftdによる分散力補正考慮)がいずれのタスクにおいても一貫して優れた精度を示すことを確認しました。特にPFPはエネルギー予測、構造最適化の安定性、比熱評価においてトップの性能を示しました。計算速度に着目すると、文献[1]の報告結果と弊社が取得した速度ベンチマークの結果からPFPがeSEN-OAMに比べ大幅に高速計算が可能であることが示唆されました。また、最新の汎用MLIPであるUMA(uma-s-1p1 task_name=odac)と比較すると、今回の検証範囲では精度、速度ともにPFPが優位であることが示されました。
以上のことから、PFPはMOF材料を探索する上で高精度・高速計算を両立しており、既存のMLIPと比較しても非常に実用性に優れたモデルであると結論できます。今年のノーベル化学賞に象徴されるように、MOFは持続可能な社会を実現する上で非常に重要な材料です。MOFの材料開発、さらには社会への貢献を実現するために、我々はMatlantisの一層の技術開発を進めて行きます。
参考文献
[1] https://arxiv.org/abs/2507.11806
[2] https://github.com/pfnet-research/torch-dftd
[3] https://matlantis.com/ja/product/about-pfp/
[4] https://arxiv.org/abs/2506.23971
[5] https://github.com/Andrew-S-Rosen/QMOF?tab=readme-ov-file
[6] https://chemrxiv.org/engage/chemrxiv/article-details/6759b06df9980725cfbc8cef
-




 URLを
URLを
コピーしました
新着記事
NEW
AIが切り拓く計算化学による材料開発

DFT 分子動力学 機械学習ポテンシャル 解説記事

NEW
京大・福井謙一記念研究センターで学ぶ、研究を加速するAI材料シミュレーション──ENEOSと共に「最高のCO2吸着剤」設計に挑む

【解説】AIはなぜそう予測したのか? PFP descriptorsとShapley値で解き明かす原子レベルの解釈性
マテリアルズインフォマティクス 解説記事 計算化学

ゼロから書くSMILES記法
解説記事 計算化学

名古屋大学×Matlantis「最先端理工学実験」レポート AIシミュレーションが実験系学生の探究心に火をつけた4日間の集中講義
インタビュー 計算化学
