材料、製薬などの研究開発の現場では、実験や評価にかかる手間やコスト、人材確保の難しさといった課題が深刻化しています。特に、人の経験や勘に頼る属人的な従来の研究開発手法では、開発スピードと精度の両立に限界が見え始めています。
こうした解題の解決方法の1つとして、マテリアルズインフォマティクス(MI)があります。MIは、実験やシミュレーションで得られたデータを機械学習モデルに学習させ、物性予測や材料探索を加速させるデータ駆動型のアプローチです。データから帰納的に推論するので、メカニズムが未解明の複雑な現象にも適用できる点が大きな強みです。
本記事では、MIの基本的な考え方から、実際に使われている技術を整理します。また、近年では、MIと計算化学との融合も進んでおり、その技術の1つである機械学習を用いてシミュレーションを高速化する技術である機械学習原子間ポテンシャル(MLIP)についても紹介します。
マテリアルズインフォマティクス(MI)とは?
これまでの材料開発は、研究者の経験や勘に頼ることが多く、属人的で多大な時間とコストを要していました。マテリアルズインフォマティクス(MI)は、データを蓄積し、そのデータをAI技術を用いて解析することによって、勘や経験の世界から脱却し、材料開発を持続的かつ効率的なものに変えるアプローチです。国内外の多くの研究機関や企業で導入が進んでいます。
MIは、機械学習モデルが材料特性を予測し、有望な材料候補を効率的に絞り込むことで、実験回数を劇的に減らせる可能性があります。これにより、開発のスピードと精度の向上が期待できます。
MIの研究開発には、マサチューセッツ工科大学(MIT)や産業技術総合研究所(産総研)、物質・材料研究機構(NIMS)といったトップレベルの研究機関や多くの化学メーカーに加え、IT企業も参入してきており、その動きは世界的に加速しています。また、技術の目覚ましい進化を背景に、具体的な成果も次々と生まれており、その可能性はますます広がっています。
MIの2大活用法:「予測」と「探索」
MIの主要な活用法は、大きく「予測」と「探索」の2つに分けられます。それぞれの特徴と使い分けを理解することが重要です。
(1)機械学習による物性「予測」
「予測」は、既知の材料データ(化学構造、製造条件など)と、それに対応する物性値(硬さ、融点、導電率など)の関係を機械学習モデルに学習させ、そのモデルを使うことで、他の化学構造や製造条件のときの物性を実験なしで予測するアプローチです。このアプローチでは、過去の膨大な実験データを有効活用できます。
機械学習モデルとしては、線形回帰や線形回帰を発展させたRidge回帰、Lasso回帰、部分最小二乗法(PLS)、カーネル法を用いるサポートベクターマシン(SVM)、木構造で予測する決定木とそれを発展させたランダムフォレストや勾配決定木、ニューラルネットワークなどデータの特徴に合わせて様々なモデルが用いられます。
(2)ベイズ最適化による効率的な「探索」
機械学習モデルはどんなデータに対しても精度良く予測できるわけではなく、学習したデータから似たデータ(内挿)に対してのみ精度良く予測ができます。そのため、手元にデータが少ない場合や、既存の材料を超える特性を探したい場合、「予測」だけでは限界があります。そこで用いられるのが「探索」のアプローチです。
「探索」のアプローチでは、”予測平均”(予測値)と”予測標準偏差”(予測の不確実性)を考慮して、どの条件で実験するかを判断します。より良い物性を達成しやすい化学構造や実験条件を計算し、その条件で実験し、結果をデータセットに加えて、モデルを再学習し、もう一度、より良い物性を達成しやすい化学構造や実験条件を計算します。これを繰り返すことで良い物性をもつ化学構造や実験条件を効率的に見つけることができます。
このアプローチはベイズ最適化と呼ばれます。ベイズ最適化では、予測値と予測標準偏差を一度に計算できるガウス過程回帰がよく用いられます。また、複数のモデル(例えば、異なるハイパーパラメータや訓練データで学習させたモデル)を用意して、それらの予測値から予測平均と予測標準偏差を計算する方法もあります。
「予測」のアプローチでは、どの機械学習モデルを選ぶかが大事でしたが、「探索」のアプローチでは、どの機械学習モデルを選ぶかに加えて、どのように探索するかも重要です。どのように探索するかは、使用する「獲得関数」によって決まります。獲得関数はいくつも提案されており、これまでの最良値より改善する確率を計算するProbability of Improvement(PI)、これまでの最良値からの改善量の期待値を計算するExpected Improvement(EI)、予測平均に予測標準偏差の定数倍を足したUpper Confidence Band(UCB)などが良く用いられます。
獲得関数の設計によって、これまでのデータの傾向を重視して活用するフェーズとこれまで評価したことない条件を重視する探索のフェーズのバランスをコントロールすることができます。1つのプロジェクトで最初から最後まで同じ獲得関数を使うこともあれば、獲得関数を切り替えながら使用することもあります。
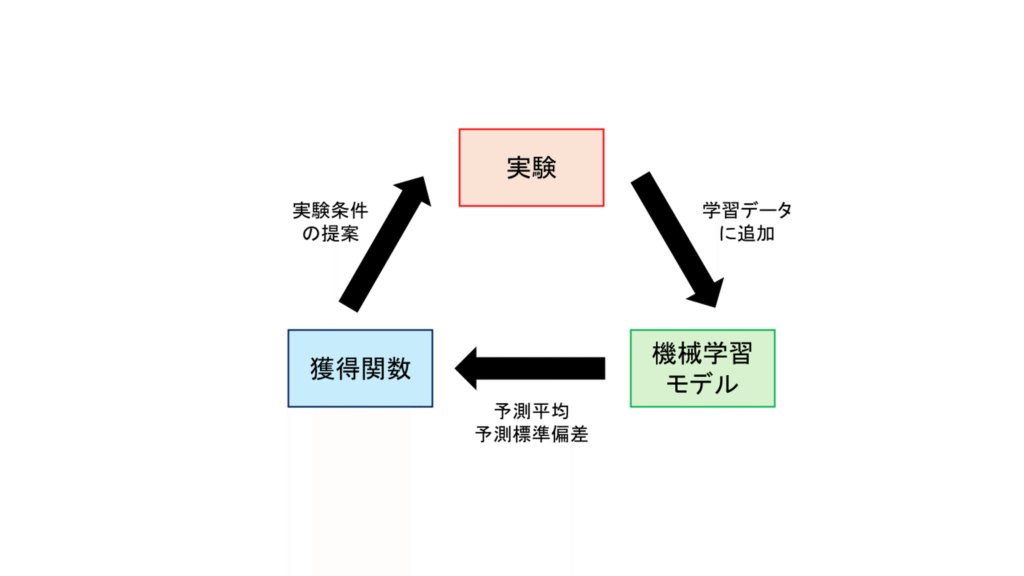
図 1. ベイズ最適化の概念図
化合物の機械学習モデルへの入力方法(化合物の特徴量の作り方)
化学分野で機械学習を利用するには、まず化学構造などの情報をコンピューターが理解できる数値データ、すなわち「特徴量」に変換する必要があります。ここでは、この特徴量を具体的にどのように作成するのかを紹介します。
(1)化学知識による特徴量抽出
化合物から特徴量を作成する方法として、化学知識を活用する手法があります。有機分子であれば、分子量や置換基の数など、無機材料では各原子の原子半径や電気陰性度の平均や分散といった情報が使われます。どのような特徴量を用いるかは人が経験や知識によって選定し、機械学習モデルに入力します。過去の知見に基づいた意味づけされた特徴量を用いることで、少ないデータでも安定した予測精度が得られるという利点があります。一方、材料の種類や目的とする物性ごとに重要な特徴量が異なるため、目的が変わる度にどのような特徴量を用いるかを見直す必要があります。
(2)ニューラルネットワークによる特徴量抽出
近年では、ニューラルネットワークにより特徴量を自動で抽出する手法に注目が集まっています。特に「グラフニューラルネットワーク(GNN)」は、原子を頂点(ノード)、結合を辺(エッジ)と見ることで分子や結晶をグラフとして扱います。図1に4-アミノフェノールをグラフとして見た図を示します。各原子は異なる特徴量を持つ頂点として表現され、結合がある原子同士の間には辺を持つようなグラフ構造になります。
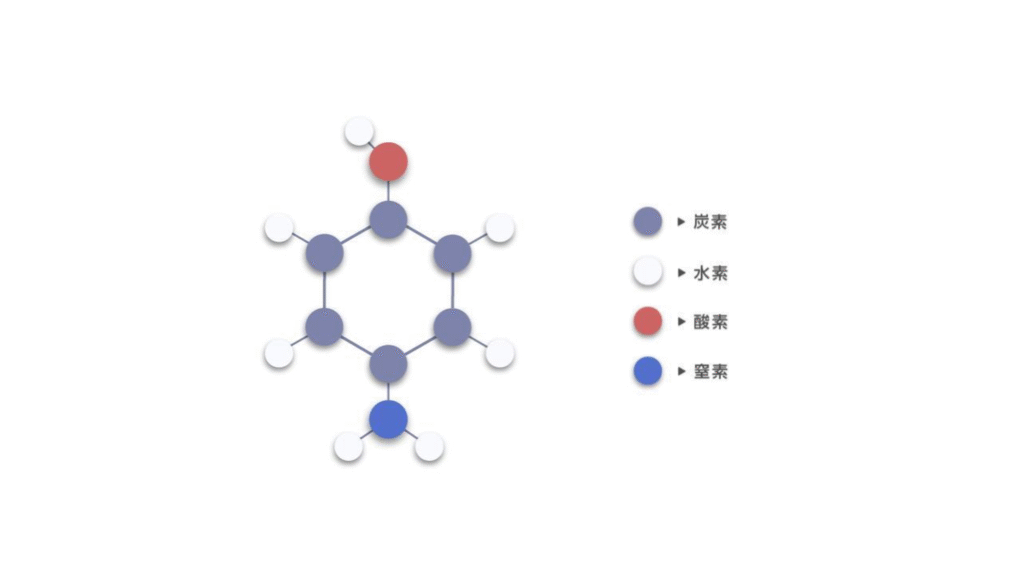
図 2. 4-アミノフェノールのグラフ構造
GNNでは、結合で繋がっている原子との位置関係や結合関係の情報を反映した特徴量を自動的に学習し、特性を予測できます。これにより、事前に有効な特徴量を設計するのが難しい場合でも、データから機械学習モデルが直接有効な情報を引き出し、高い予測精度の実現につなげることができます。ただし、一般的には、適切な学習には多くのデータを必要とします。
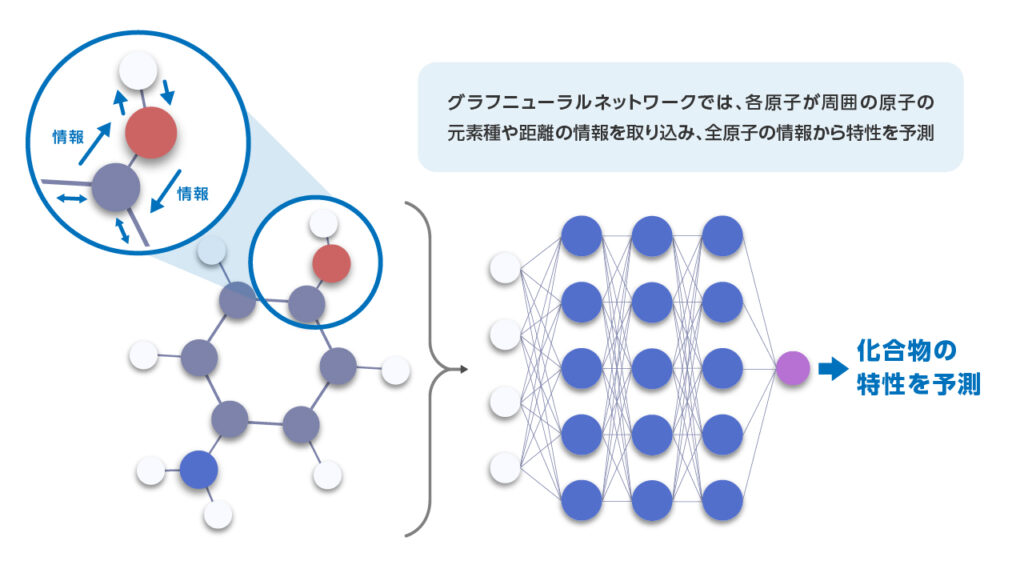
図 3. GNNを用いた4-アミノフェノールの特性予測
MIと計算化学の融合:データ不足を克服する新潮流
MIでは、機械学習モデルを学習させるためのデータは、実験を行って取得するため、取得コストが高い場合が多く、学習に使えるデータが限られてしまうことがあります。この課題への対応策の1つとして、計算化学との融合があります。
計算化学との融合には、大きく分けて2つの方向性があります。1つは、量子化学計算などから計算した物性を化合物の特徴量として用いることで、機械学習モデルの精度を向上させるという機械学習モデルの精度向上させる従来からのアプローチです。
そしてもう1つが、近年特に注目されている、機械学習で計算化学を高速化する技術、機械学習原子間ポテンシャル(MLIP)です。
材料の物性を高い精度で予測するには、DFT計算で原子間の相互作用を求め、分子動力学(MD)シミュレーションを行うのが有効です。しかし、この手法は膨大な計算時間を要するという大きな壁がありました。MLIPは、この原子間の相互作用を機械学習でモデル化したもので、DFT計算と同等の精度を保ちながら、計算速度を数十万倍以上にまで劇的に向上させることができます。
これにより、これまで時間がかかりすぎていた多様な構造や条件でのシミュレーションが迅速に行えるようになり、実験前のスクリーニング精度が飛躍的に向上します。さらに、シミュレーションで得られた膨大なデータをMIの学習データとして活用することで、データ不足の課題そのものを解消し、内挿を大きく広げる非常に有望なアプローチです。
まとめと今後の発展について
本記事では、マテリアルズインフォマティクス(MI)の基本から応用までを解説しました。MIは、単なるツールの導入に留まらず、研究開発のあり方そのものを、経験と勘に基づく従来型からデータ駆動型へと変革するアプローチです。
MIの核心は、AI・機械学習を用いて、材料特性の「予測」や、最適な条件の効率的な「探索」を可能にすることです。一方で、課題としてデータ不足が挙げられますが、これに対してはMLIPが解決方法の1つになります。MLIPを用いることで、高速かつ高精度なシミュレーションが可能となり、実験データが一つもなくても材料の特性を予測できるようになります。
今後は、ロボティクスによる実験の自動化、ChatGPTに代表される大規模言語モデル(LLM)による論文や実験ノートなどのテキスト情報から構造化データへ変換によって、データ量の課題は解決され、MIが材料開発を一層加速させると期待されます。
-




 URLを
URLを
コピーしました
新着記事
NEW
AIが切り拓く計算化学による材料開発

DFT 分子動力学 機械学習ポテンシャル 解説記事

NEW
京大・福井謙一記念研究センターで学ぶ、研究を加速するAI材料シミュレーション──ENEOSと共に「最高のCO2吸着剤」設計に挑む

【解説】AIはなぜそう予測したのか? PFP descriptorsとShapley値で解き明かす原子レベルの解釈性
マテリアルズインフォマティクス 解説記事 計算化学

ゼロから書くSMILES記法
解説記事 計算化学

名古屋大学×Matlantis「最先端理工学実験」レポート AIシミュレーションが実験系学生の探究心に火をつけた4日間の集中講義
インタビュー 計算化学
