Monolayer to bilayer phase transformation of CaSi 2Fx (0 <x<1.5)
Theme Overview
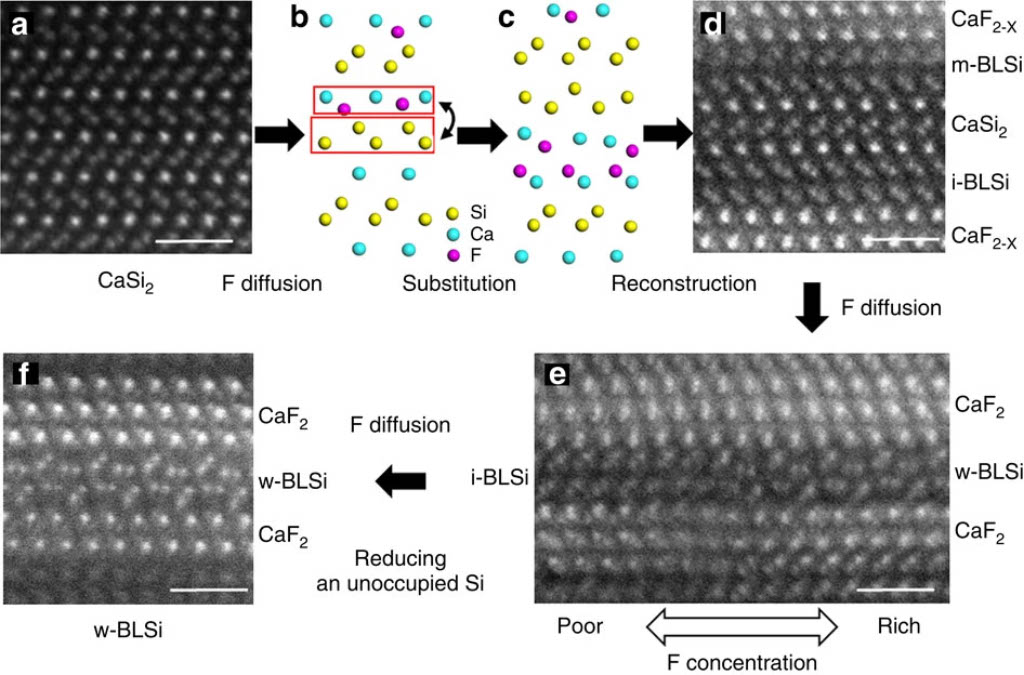
CaSi2 consists of alternatively stacked structures of Ca and Si monoatomic layers, and it is used as a raw material for silicene compounds. [1] By fluorine diffusion into CaSi2, the crystal structure transforms to bi-layered and multi-layered structures.
The reactions accompanying the insertion of atoms into such layered materials are important for the delamination and control of sheet materials, as well as for electrode materials in secondary batteries, etc. On the other hand, it is necessary to investigate the structural stability of the composition, which has been studied by fitting cluster expansions to DFT results[2].
In this case study, the results of DFT literature were reproduced using PFP, and the stability of structures not found in the literature was also evaluated. As a result, the structural phase transition corresponding to the experiment was confirmed by calculation. Furthermore, the reaction dynamics was also analyzed by performing large-scale MD simulations with over a thousand atoms.
Calculation Models and Methods
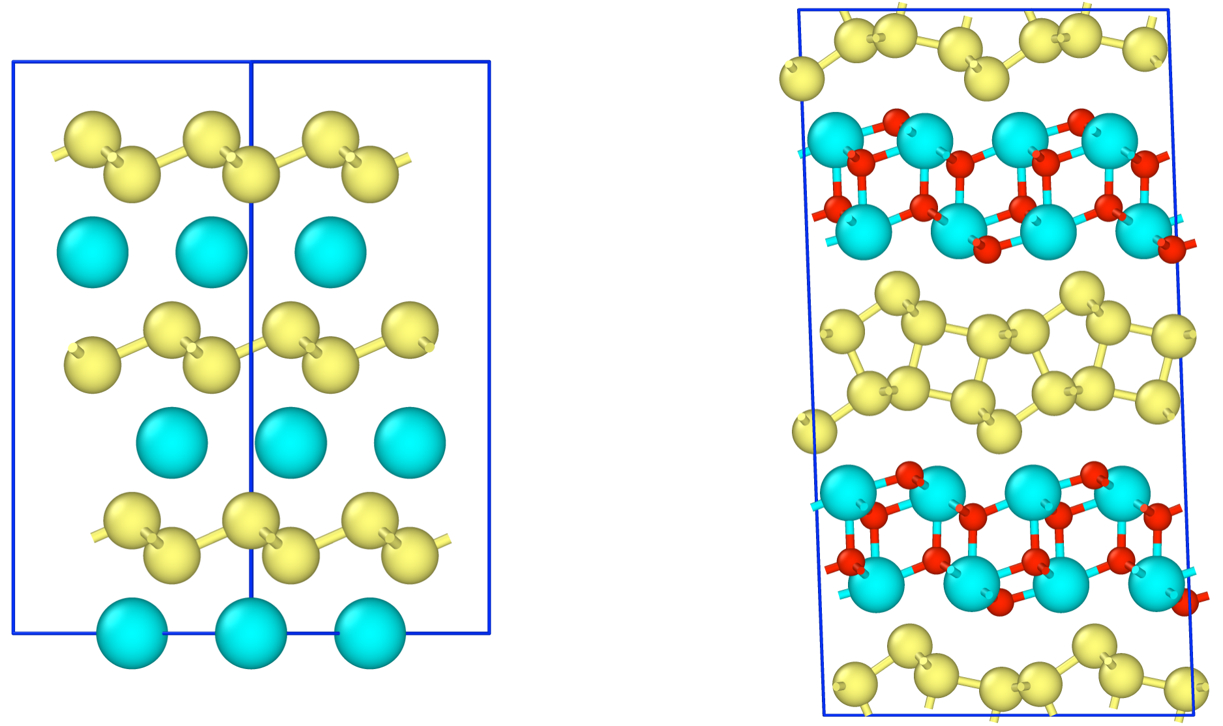
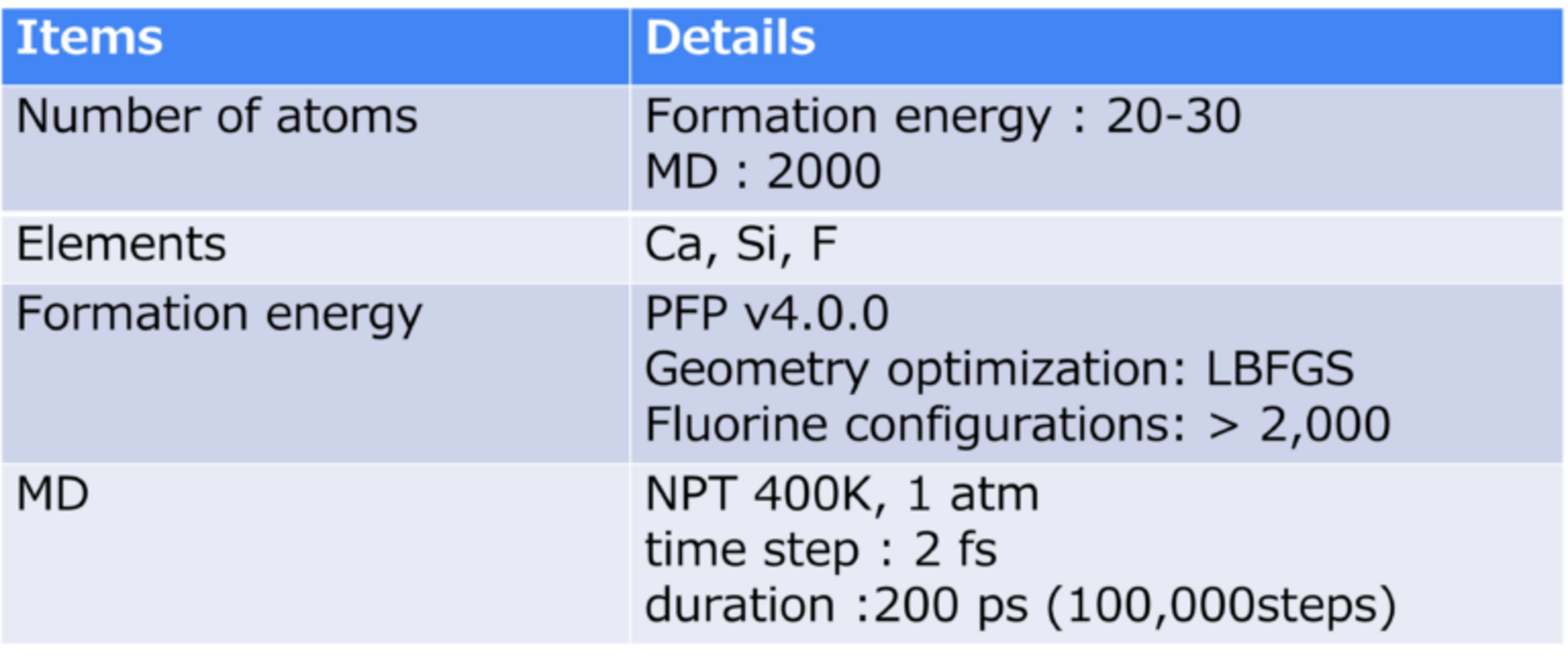
The formation energy of CaSi2Fx (0<=x<=1.5) was calculated using PFP with respect to the Ca/F composition ratio x to obtain stable structures. In addition to the monolayer CaSi2, we also performed calculations for the bilayer structures (i-bilayer and wavy-bilayer) observed in experiments.
The python package icet was used to search for stable F configurations [3]. The formation energies of all the structures of various F configurations generated by icet using PFP were evaluated.
Results and Discussion
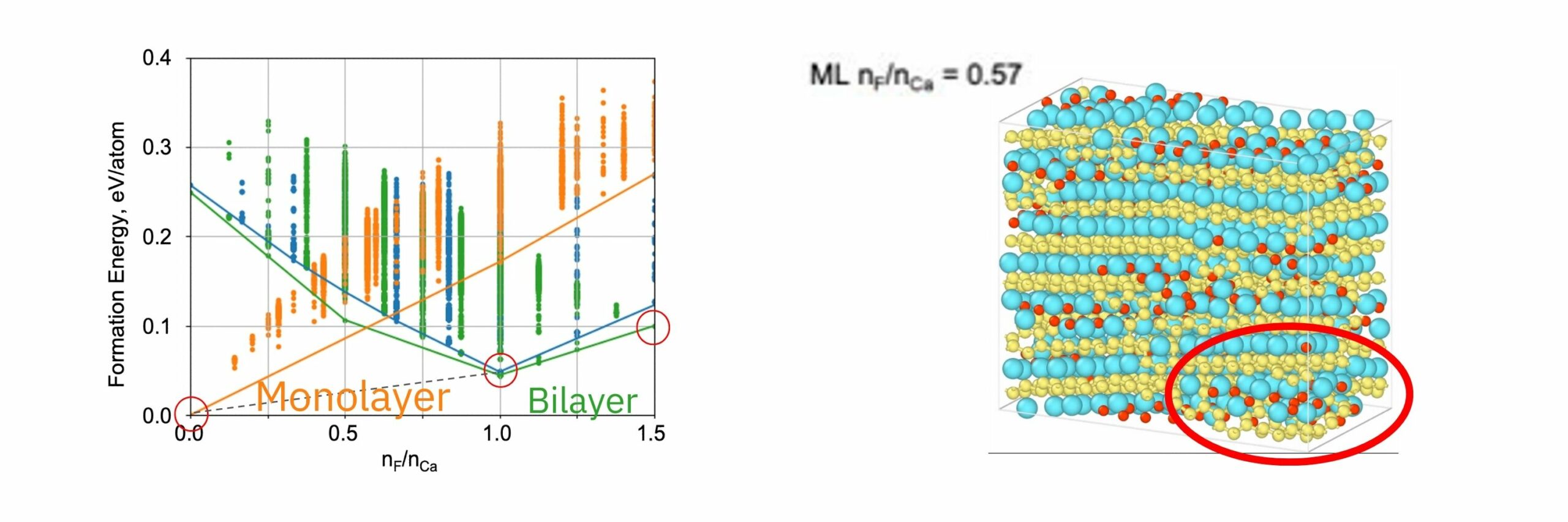
The formation energy calculations showed that the bilayer structure is more stable than the monolayer structure at F/Ca ratios of 0.5 and above, which is consistent with the DFT literature. At F/Ca ratios of 1.0, the i-bilayer structure is stable, and at 1.5, the wavy-bilayer structure is stable. The calculation results were quantitatively consistent with the results of the structural phase transition observed by STEM.
In the MD simulations, when F is inserted into CaSi2, a two-atom layer structure of CaF2 was formed in the region of high F density, and then a structural phase transition occurs as Si diffuses between the layers.
These results confirmed that PFP calculations can provide results consistent with experiments for calculations involving changes in the charge balance, such as the insertion of ionic atoms between the lattice of a crystalline material.
Computational Details
References
[1] R. Yaokawa, Nat. Commun. 7, 10657 (2016). [2] A. Nagoya, Phys. Chem. Chem. Phys., 23, 9315 (2021) [3] icet. https://icet.materialsmodeling.orgtag
Publication date of this case: 2024.03.26
-




 URL
URL
Copied
what's new
NEW
High-Accuracy, High-Speed Binding Energy Evaluation of Cyclin-Dependent Kinase 2 Inhibitors
Pharmaceuticals
NEW
Data-Driven Method for Discovering Low Adsorption Energy Molecules on Si Surfaces
ScreeningMaterials InformaticsSemiconductors
Calculation and Verification of Methanol Synthesis Catalysts
Methanol synthesis catalystsLarge-scale screening
Direct derivation of atomic displacement parameters using NNP-MD
Atomic Displacement ParametersThermoelectric Materials

A Statistical Understanding of Oxygen Vacancies in High-Entropy Perovskite Oxides
CeramicsHigh Entropy MaterialsElectrolysis
