

材料開発をはじめとする研究開発の現場では、実験にかかる手間やコスト、人材確保などの課題があります。こうした中、計算化学によるシミュレーションを活用したアプローチが注目されています。特に、量子力学に基づいて電子状態を計算するDFT(Density Functional Theory:密度汎関数理論)は、計算コストと精度のバランスに優れ、研究開発現場での活用が広がりつつあります。
本記事では、DFT計算を実施するための代表的なソフトウェアについて、選定ポイントや対応可視化ツール、そして実際の計算の流れをわかりやすく解説します。また最後に、当社製品であるMatlantisを利用した高速計算との比較も簡単にご紹介します。
DFTソフトウェアの選択ガイド
DFT計算ができるソフトウェアにはさまざまな種類があり、期待する計算結果を得るためには、目的にあったソフトを導入する必要があります。ここでは、DFTソフトウェアを選定する際の基本的なポイントとして、対象とする物質や見たい物性、利用環境や予算などについて解説します。
対象とする物質系を明確にする
DFT計算では対象とする物質系によって、標準的に用いられる計算方法が異なります。そのため、対象とする物質が「固体系」か「分子系」かを、あらかじめ整理しておく必要があります。大まかには以下のように分類できます。
「固体系」とは、金属や半導体などの固体や、アモルファスや溶液などの凝集構造が該当します。入力構造が周期的に連なった構造を想定することで(周期境界条件)、無限に大きな系に対する計算が可能です。原子・分子の集合体としての電子状態や物性を計算したい場合には、固体系向けDFTソフトウェアを選択してください。
「分子系」とは、文字通り水(H₂O)やメタン(CH₄)などの分子や、それらが相互作用したクラスターなどが該当します。通常真空中での計算になりますが、溶媒効果を考慮した計算も可能です。分子単体での性質や、均一系の反応を高精度に計算したい場合などには、分子系向けDFTソフトウェアの方が適しています。
見たい物性・現象を整理する
次に、計算したい物性を整理します。基本的には、電子状態が関係する物性であれば、DFT計算による評価が可能です。
計算によって得られた物性は、実験データと比較して妥当性を評価することが重要です。ソフトによって対応できる物性が異なるため、あらかじめ取得可能な実験データに対応しているかを確認しておくことが大切です。
以下に、DFTの計算対象となる主な特性・現象と、得られる計算結果の代表的な例を示します。
| 計算対象の特性・現象 | 得られる計算結果の例 |
| 構造特性 | 格子定数、表面構造、平衡構造、体積、密度 |
| 電子的特性 | バンド構造、状態密度、分子軌道、部分電荷、双極子モーメント |
| 熱力学的特性 | 比熱、熱容量、沸点、融点、形成エネルギー、表面エネルギー、自由エネルギー |
| 輸送特性 | 電気伝導度、拡散係数、熱伝導率、粘度 |
| 応答関数・光学特性 | 弾性定数、誘電率、磁気モーメント、フォノン、分子振動、紫外可視吸収波長・強度 |
| 化学反応 | 反応エネルギー、活性化エネルギー |
電子状態と関係する物性であれば、原理的には多くの実験物性に対応可能ですが、対象とする時間・空間スケールによっては、実用的でない場合もある点に留意が必要です。現実的にDFT計算で対応可能な時間・空間のスケールの上限は、それぞれns・nmのオーダーです。
ビューアーについて確認する
DFTソフトウェアで計算を実施する際には、入力として計算対象の三次元座標が必要です。公開データベースや論文等から構造を取得できる時もありますが、もし手に入らない場合には構造の作成(モデリング)を行う必要があります。またDFT計算の結果は、一般的に数値中心のテキスト形式で出力されます。計算内容によっては数万行に及ぶこともあり、特に初学者にとっては、どこに欲しい情報が記載されているのかを見つけることが困難です。
そのため、「ビューアー」とよばれる可視化ソフトが利用されます。ビューアーがモデリング機能を備えている場合も多く、複雑な分子や表面吸着系などについては、この機能を使って作成することが一般的です。ソフト対応の公式ビューアーであれば、エネルギーや電荷などの情報を、出力ファイルから取得・表示してくれます。また構造、分子軌道、振動など、数値のままでは理解しにくい情報の可視化には特に有効です。
ビューアーにはさまざまな種類があり、使用するDFTソフトウェアに対応したものを選ぶ必要があります。可視化できる物性、機能、操作性、サポート体制、有償無償などもビューアーによって異なるため、導入前に確認しておくことが重要です。
利用可能な環境・リソースを把握する
DFTソフトウェアで計算を行うには、使用するパソコンや計算機環境など、利用可能なリソースの把握が重要です。小規模な計算であれば汎用のパソコンでも実施可能ですが、本格的な運用には専用計算機の利用が一般的です。
ただし、専用計算機を新たに導入する場合、高額な初期費用に加え、設置場所の確保、電力や冷却などの環境整備、さらにシステム構築や保守管理に関する専門知識・人員も必要となります。
また、計算対象の原子数(電子数)が多い場合や、高精度な結果を求める場合には、計算量が大幅に増加し、1回の計算に数週間かかることもあります。そのため、実行できる試行回数が限られ、計算リソースの制約が研究開発の大きな課題となることも少なくありません。
このため近年では、クラウドサービスを利用して計算環境を確保する方法や、AIを活用して計算を高速化する技術も登場しており、計算リソースに対する選択肢の幅が広がっています。例えば、当社が提供するクラウドサービス「Matlantis」は、インフラ構築不要で導入でき、機械学習技術を応用した高速計算が可能なソリューションとして、多くの現場でご活用いただいています。
予算・ライセンス形態を検討する
DFTソフトウェアを選定する際は、導入費用やライセンス形態も重要な検討項目となります。
ソフトには、オープンソース(無償)と商用(有償)のものがあり、無償ソフトには機能やサポート、ライセンス形態に制限がある場合があります。一方、有償ソフトでは、アカデミック用途で数十万から数百万円程度が一般的ですが、産業用途ではその10倍以上の費用が発生することもあります。
ライセンス形態もさまざまで、個人単位での使用を前提とした「ネームドライセンス」、特定の端末に限定される「ノードロック」、組織内で複数人が利用できる「サイトライセンス」などがあり、ソフトによって条件は異なります。また、ソフトを購入せず、必要な時に計算を委託できる「受託計算サービス」を活用する方法もあります。
主要なDFTソフトウェア比較
以下に、主要なDFTソフトウェアの一覧を示します。各ソフトの主な特徴、対応するビューアー、ライセンスの有償無償について簡単に整理しています。なお、仕様の詳細や最新情報については、各ソフトの開発元の公式サイトをご確認ください。導入の際は初期設定や解析時のサポート体制なども事前に確認しておくと安心です。
| ソフトウェア名 | 主な対象系 | 主な特徴 | 主な対応ビューアー | ライセンス |
| VASP | 固体系 | 固体・周期系計算の業界標準 |
p4vasp VESTA | 有償 |
| Quantum Espresso | 固体系 | 無償で利用できる固体・周期系計算ソフトウェア | VESTA | 無償 |
| SIESTA | 固体系 | 目的に合わせて、電子の数学的表現を調整可能 | VESTA | 無償 |
| Gaussian | 分子系 | 分子系計算の業界標準、GUI版もあり |
GaussView Avogadro | 有償 |
| GAMESS | 分子系 | 無償・機能開発が盛ん |
MacMolPlt Avogadro | 無償 |
| ORCA | 分子系 | 光学物性系や高精度計算に強み |
Avogadro ChimeraX Chemcraft | 有償(アカデミック無償) |
・固体材料向け
固体系に対応したソフトには、VASP、Quantum Espresso、SIESTAなどがあります。VASPは有償ソフトで、固体系計算の業界標準として広く利用されています。Quantum EspressoとSIESTAは無償で提供されており、アカデミック用途を中心に活用されています。対応するビューアーとしてはVESTAやp4vaspがあります。計算結果の構造をcifファイル(結晶構造向けの形式)やxyzファイル(原子数、元素種、三次元座標のみからなるシンプルな形式)に変換すれば、結晶構造向けのソフトで可視化することも可能です。
・分子材料向け
分子系に対応したソフトには、Gaussian、GAMESS、ORCAなどがあります。Gaussianは有償ソフトで、分子系計算の業界標準として知られています。GAMESSは無償で提供されており、機能開発が活発に行われています。ORCAは有償ソフトですが、アカデミックユーザー向けには無償で提供されています。対応するビューアーには、GaussView(有償)、Avogadro、Chemcraftなどがあります。
Python上で動くDFT計算向けライブラリ
専用計算機を新規に導入し、本格的に計算を開始したい場合には、通常Linux環境でのコマンドライン操作やコンパイル(=プログラムを計算機が実行可能な形式に変換)が必要です。これらの作業に不慣れな場合、環境構築の時点で躓いてしまい、別途コストがかかる可能性があります。
こうした中、DFT計算が可能なPythonライブラリ(=開発済みプログラム集)が公開されており、PySCFやPsi4がその代表例です。Pythonは機械学習の分野でも広く利用されており、すでに業務へ活用されている方も多い状況かと思います。Pythonコードベースで構造作成や計算条件設定が可能であるため、Pythonに慣れたユーザーにとっては、導入・計算実行のハードルが低い選択肢の一つです。
様々なPythonライブラリを組み合わせることによって、材料特性に関する計算データ取得、機械学習・データ解析への流れを、シームレスに実行できます。そのため、近年注目されているマテリアルズ・インフォマティクス(MI)の実施にあたって、非常に相性の良い環境を構築することが可能と言えます。
DFT計算を始めるための実践的ステップ
DFTソフトウェアの選び方や主要ソフトの特徴を踏まえ、ここでは実際にDFT計算を実施する際の基本的なステップを解説します。使用するソフトによって差はあるものの、大まかな流れを把握する参考としてご活用ください。
ステップ1:計算機環境を整える
まず、使用するDFTソフトウェアやビューアーを、公式サイトのマニュアルなどに従って、パソコンまたは専用計算機にインストールします。
CPUのコア数が多いほど並列計算が可能になり、処理速度が向上します。専用計算機では、数十から数百コアのCPUが用いられることもあります。またGPUを搭載して高速化を図ることも可能です。
計算対象の原子数が数十を超える規模であるなど、計算量が増大する場合には、より高性能な計算機環境を整える必要があります。クラウド上でDFT計算を実行する環境も、選択肢の一つになります。
ステップ2:入力ファイルを作成する
DFT計算を行うには、あらかじめ「入力ファイル」を作成しておく必要があります。これは、計算対象の物質の情報と計算方法を記述したテキストファイルであり、DFTソフトウェアに読み込ませることで計算が実行されます。
具体的には、三次元構造(原子の配置)、電荷、スピンなどの計算対象の情報に加え、求めたい物性(エネルギー、安定構造、振動など)や計算条件(汎関数、基底関数など)を記載します。
入力ファイルは、テキスト形式で直接記述するほか、ビューアーやモデリングソフトのGUIから作成することも可能です。
以下に、GAMESSでアスピリンの構造最適化計算を実行する際の入力ファイル例を示します。アスピリンの三次元構造は、公開データベースのPubChem(https://pubchem.ncbi.nlm.nih.gov/)から取得しています。また、このインプットはAvogadro2(https://www.openchemistry.org/projects/avogadro2/)というビューアーの機能を使って作成しました。
! Aspirin opt
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 $END
$CONTRL SCFTYP=RHF RUNTYP=OPTIMIZE ICHARG=0 MULT=1 DFTTYP=WB97X-D $END
$STATPT OPTTOL=0.0001 NSTEP=100 $END
$SYSTEM MWORDS=2 $END
$DATA
Aspirin
C1
O 8.0 1.23330 0.55400 0.77920
O 8.0 -0.69520 -2.71480 -0.75020
O 8.0 0.79580 -2.18430 0.86850
O 8.0 1.78130 0.81050 -1.48210
C 6.0 -0.08570 0.60880 0.44030
C 6.0 -0.79270 -0.55150 0.12440
C 6.0 -0.72880 1.84640 0.41330
C 6.0 -2.14260 -0.47410 -0.21840
C 6.0 -2.07870 1.92380 0.07060
C 6.0 -2.78550 0.76360 -0.24530
C 6.0 -0.14090 -1.85360 0.14770
C 6.0 2.10940 0.67150 -0.31130
C 6.0 3.53050 0.59960 0.16350
H 1.0 -0.18510 2.75450 0.65930
H 1.0 -2.72470 -1.36050 -0.45640
H 1.0 -2.57970 2.88720 0.05060
H 1.0 -3.83740 0.82380 -0.50900
H 1.0 3.72900 1.41840 0.85930
H 1.0 4.20450 0.69690 -0.69240
H 1.0 3.71050 -0.36590 0.64260
H 1.0 -0.25550 -3.59160 -0.73370
$END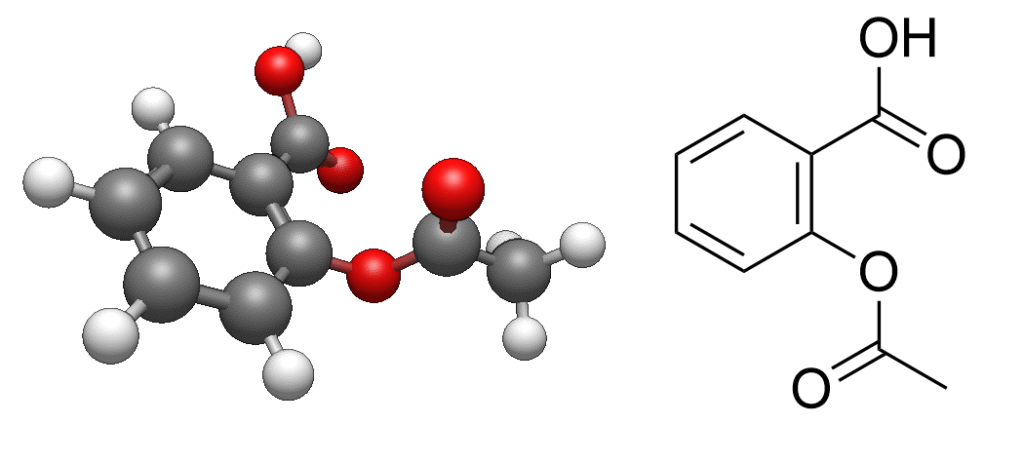
ステップ3:計算を実行し、出力結果を確認する
DFT計算を実行すると、結果が「出力ファイル」として生成され、数値を中心としたテキスト形式で出力されます。
例えば、ステップ2で作成した入力ファイルを使ってアスピリンの構造最適化計算を行うと、原子位置が段階的に変化し、エネルギーの低い安定構造へと収束します。この過程での構造やエネルギーが、出力ファイルに記録されます。
こうした出力結果は、前述のビューアーを使用することで可視化でき、解析も容易になります。GAMESSの出力ファイルをAvogadro2に読み込ませると、以下のような構造最適化後の構造が表示されます。
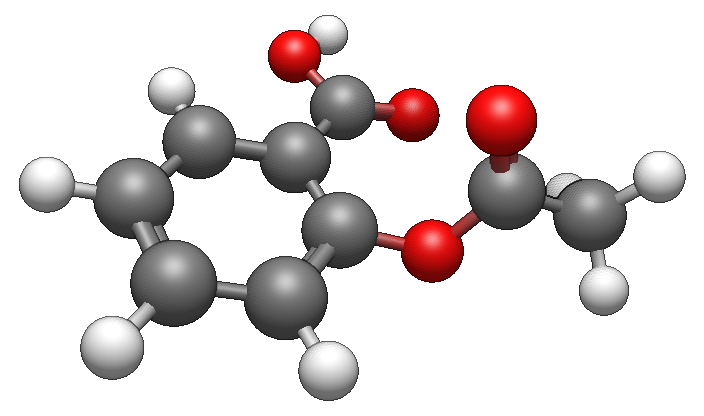
構造最適化の結果、特にカルボキシル基(-COOH)が芳香環と同じ面に来るように構造変化しています。このような計算によって、構造式からは見えない、分子の立体的な構造に関する情報が得られることがわかります。通常、分子は安定構造で存在しているため、DFT計算ではまず構造最適化を行い、得られた安定構造を基に電子状態・物性について議論するのが一般的です。
この計算を通常の作業向けノートパソコン(11th Gen Intel(R) Core(TM) i7-1165G7 @ 2.80GHz)で実行した際には、約6,400秒の計算時間がかかりました。また、同じ計算を複数CPUで処理する並列計算を行った場合では、約4,300秒/2コア・約3,900秒/4コアでした。
Matlantisを用いた計算との比較
当社製品のMatlantisでは、上の例と同じDFT計算(汎関数・基底関数)の結果を再現できる、機械学習モデルを提供しております。同様の構造最適化計算をMatlnatisで実行した結果、計算は約9秒で完了しました。計算機の条件が異なるため、直接比較はできませんが、Matlantisによる計算の速度感がよく分かるかと思います。
一般的なDFT計算では、系の電子数の3乗オーダーで計算量が増加するため、原子数が増えるほどMatlantisによる高速化が顕著になります。そのため、Matlantisでは、DFT相当の計算を約10万倍(256原子系)から約2,000万倍(3000原子)早く実行することできます(詳細は弊社HP内のProductをご参照ください)。したがって、従来のDFT計算では不可能だった原子数の取り扱いや、MI向けハイスループット計算など、時間が課題になっていた領域への展開が可能になります。
なお、Matlantisはクラウドサービスであるため、ユーザー側で環境構築・メンテナンスの手間は不要です。また、 Webブラウザで動作するアプリケーションである「Jupyter Notebook」上で、種々のPythonコードやライブラリを活用して作業をする設計になっています。そのため、計算はもとより、構造作成・可視化、解析や機械学習などの作業を、Matlantis内だけで完結させることが可能です。
まとめ
本記事では、DFTソフトウェアの選び方、主要ソフトの比較、導入ステップについて解説しました。 DFTソフトウェアにはさまざまな種類があり、用途や研究目的、予算、運用体制に応じて、最適なソフトを導入することが重要です。
併せて、計算環境の整備が不可欠です。計算速度を高めることで、より多くの試行が可能となり、研究開発のスピードと精度の向上につながります。本記事を参考に、ご要望に合ったDFTソフトを選択・導入いただき、DFT計算を活用した研究開発の推進に繋がれば幸いです。 当社が提供するMatlantisは、深層学習を活用した高速な汎用原子シミュレーションツールです。クラウド環境で利用でき、従来のDFT手法と比べて数オーダー以上の高速化を実現しています。興味のある方は、Matlantisの計算事例・導入資料もぜひご覧ください。
-




 URLを
URLを
コピーしました







