本記事は、技術専門誌『月刊マテリアルステージ』(2026年2月号)特集「AI,自動実験を活用した材料開発の効率化」に掲載された、Matlantis株式会社 カスタマーサクセスエンジニア 張による執筆記事を、ブログでも公開するものです。

1. これまでの材料開発
我々の生活は、数々の革新的な材料によって豊かになってきた。だが、その材料開発の裏には、気の遠くなるような泥臭い試行錯誤の歴史が隠されていることがある。その象徴的な例として、ハーバー・ボッシュ法が挙げられる。ハーバー・ボッシュ法は窒素と水素からアンモニアを合成する方法で、20世紀最大の発明の1つとも言われる。しかし、この技術の工業化に不可欠な触媒は、Alwin Mittaschが約2500種類について実験し、ようやく発見したものである[1]。
このような背景からもわかるように、確かに材料開発は実験主導の側面が強いと捉えられがちである。しかし、理論的アプローチもまた重要な役割を担ってきた。密度汎関数理論(DFT)に代表される第一原理計算は材料のエネルギーや各原子に働く力、電子状態などを算出できる。これらを用いることで、材料の安定構造の特定をはじめ、原子レベルのダイナミクス解析や化学反応経路の探索も可能となる。本稿では、これらの手法を用いた研究アプローチを「計算化学」と呼ぶ。計算化学は、実験に先立って物質の特性を予測し、有望な候補を絞り込むことで、開発プロセスを大幅に効率化する可能性を秘めている。
しかし、計算化学が材料開発の現場で常に活用されているとは言い難い。その根底には、DFTに代表される高精度な手法は計算時間が極めて長く、一方で古典ポテンシャルのような高速な手法は精度や汎用性に限界があるという、「精度と計算時間、汎用性のトレードオフ」が根深く存在する。
この膠着状態を打破する技術として、近年、機械学習(ML)が注目を集めている。自然言語処理や画像認識でブレイクスルーを巻き起こしたMLを計算化学に応用し、高精度な計算と高速な計算を両立させようという試み、それが機械学習ポテンシャル(MLIP)である。
本稿では、これまでの計算化学手法を振り返りながら、機械学習が精度と計算時間、汎用性のトレードオフをいかにして克服し、材料開発のパラダイムを根底から覆しつつあるかを詳述する。DFTの計算結果を学習し、その予測能力を維持したまま計算速度を劇的に向上させるMLIPの登場、さらに、その発展形である汎用機械学習ポテンシャル(uMLIP)が、ゲームチェンジャーとして台頭してきた現状を解説する。そして、uMLIPを用いた材料開発の最前線を追う。これは単なる計算化学の高速化ではない。AIによって材料開発プロセスを再定義する、新時代の幕開けである。
2. 従来の計算化学手法とその課題
ここまでで述べたように、計算化学が材料開発の現場で広く活用されるには「精度と計算時間、汎用性のトレードオフ」という根深い課題が存在する。本章では、まず従来の計算化学手法を代表するDFTと古典ポテンシャルを取り上げ、それぞれが抱える問題点を具体的に詳述する。
2.1 DFT
計算化学の根幹をなす主要な手法の1つとして、DFTがある。DFTは、系の電子密度を用いてエネルギーや各原子に働く力などを計算する手法である。計算精度は用いる交換相関汎関数に依存するものの、実験結果を再現する高精度な結果を与えることも可能であり、固体物質の電子物性予測や分子の安定性、反応性の評価などで活用されてきた。さらに、DFTで得られる原子に働く力を利用して、様々な解析をすることも可能である。ここでは、系の時間発展をシミュレートする分子動力学(MD)法と、化学反応の経路を探索するNudged Elastic Band(NEB)法を紹介する。
- MD法 : 各原子について古典力学の運動方程式を数値的に解くことで、原子の座標、速度、エネルギー等の時系列データを生成する。このデータを解析することで、拡散係数や動径分布関数といった物性値を算出できる。
- NEB法 : ある安定状態から別の安定状態への反応経路を予測する手法。遷移プロセス中の系の構造変化を明らかにすると同時に、反応の障壁となる活性化エネルギーを見積もることができる。
DFTとこれら手法を組み合わせることで、多様な物性を高精度に計算できる。しかし、DFTの計算時間は系の電子数の3乗以上に比例して増大するため、極めて長い。特にMD法では、シミュレーションの各時間ステップでDFT計算を行う必要があり、一般的に扱える原子数は数百、時間スケールはピコ秒程度に限られる。これでは、材料開発で重要となる多くの現象を扱うことは困難である。
2.2. 古典ポテンシャル
DFTの長い計算時間を回避するため、経験的な関数で原子間相互作用を記述する「古典ポテンシャル」も広く用いられてきた。金属材料に適用可能なEAMポテンシャル、共有結合性物質に適用可能なTersoffポテンシャル、化学反応を記述可能なReaxFFなど、対象とする物質や現象に応じて多様な古典ポテンシャルが開発されている。古典ポテンシャルを用いたMD法は非常に高速であり、数百万原子規模のシミュレーションをマイクロ秒オーダーで追跡することも可能である。しかし、その利便性と引き換えに二つの大きな制約を抱えている。第一に、精度の問題である。相互作用を単純な関数形で近似しているため、DFTと比較して予測精度が劣る。第二に、汎用性の欠如である。ポテンシャルの多くは、特定の材料系に対してパラメータが調整されており、異なる材料系には適用できない。シミュレーション対象が変わるたびにポテンシャル関数を再調整する必要があるため、網羅的な材料スクリーニングへの応用は難しい。
このように、DFTを用いる手法はその計算時間の長さから、古典ポテンシャルを用いる手法は精度と汎用性の低さから、ハイスループットな材料開発の実現を阻んできた。そのため、これまでの多くの場合、計算化学が材料開発に貢献できる機会は限定的であり、実験による検証が先行するのが実情であった。
3. 機械学習原子間ポテンシャル(MLIP)
3.1 MLIPの基本概念と歴史的ブレイクスルー
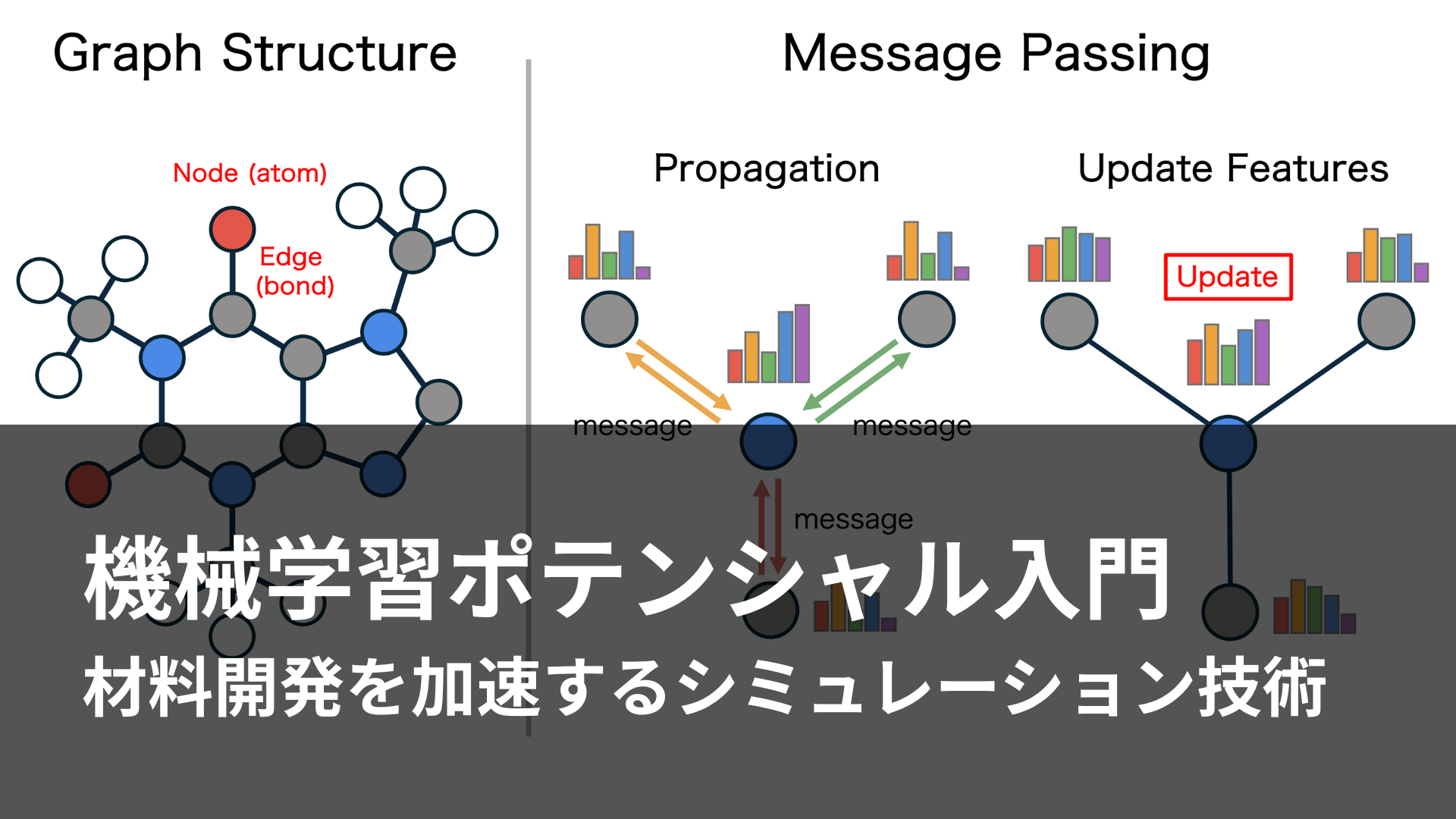
従来の計算化学が直面していた精度と効率のトレードオフを抜本的に解決するアプローチとして登場したのが、MLIPである。MLIPの基本原理は、入力(原子の種類や座標)と出力(DFT計算などで計算されたエネルギーや力)のペアを大量に用意し、その関係をMLモデルに学習させるというものである。一度学習されたモデルはDFT計算よりも遥かに高速にエネルギーや力を予測できるため、DFT計算に匹敵する精度を保ったまま、高速にMD法やNEB法を実行可能である。
MLIPの機械学習モデルには、ニューラルネットワーク(NN)が頻繁に採用される。NNは高い表現力を有するのに加え、微分と相性が良い。物理学において原子に働く力は、エネルギーを原子座標で微分することで算出され、NNはこの計算を効率的に行えるため、適しているのである。MLIPの研究は1995年には既に始まっていたが[2]、初期のモデルは、(1)扱える原子数が固定されている、(2)並進・回転対称性など物理的に満たされるべき対称性を任意の系で保証できない、といった課題を抱えていた。原子座標に対称性を考慮した変換を施すなどの改良が試みられたが[3]、ブレイクスルーは2007年に訪れた。ベーラーとパリネロが提案したMLIP[4]は、まず対称性関数を用いて各原子の局所環境を記述する。次に、パラメータを共有したサブネットが各原子のエネルギーを推論し、それらを合計することで系全体のエネルギーとする。この「系全体のエネルギーは各原子エネルギーの和」とするアーキテクチャにより、学習データと異なる原子数の系にも適用可能となった。そして、彼らは約8,200点のDFT計算結果を用いて学習させたMLIPが、溶融シリコンの動径分布関数をDFT計算と同等の精度で、かつ64原子系では数十万倍高速に計算できることを示し、MLIPが有力なシミュレーションツールとなり得ることを示した。
3.2 汎用性への挑戦とuMLIPの実現
ベーラーとパリネロはMLIPの有用性を明確に示したが、材料開発のプロセスを革新するには、まだ「汎用性」という大きな壁が残っていた。機械学習モデルは、原理的に学習データに含まれない未知の状況(外挿)の予測を苦手とする。そのため、特定の元素や原子環境で訓練されたMLIPは、異なる系ではエネルギーや力を正確に予測できず、シミュレーションが破綻してしまう。これは、古典ポテンシャルが抱えていた「対象が変わるたびにパラメータを再調整する必要がある」という問題点と本質的に同じであった。この課題の克服には、多様な原子環境を記述できる表現力の高いモデルアーキテクチャと、その学習に必要な広範なデータセットが不可欠である。この方向性での研究が、uMLIPの潮流を生み出した。2017年に発表されたANI-1では、ベーラーとパリネロが提案した手法を複数の元素に適用できるようアーキテクチャが改良され、学習データセットの構築には効率的なデータ生成手法が導入されている。これにより、H、C、N、Oの4元素に対応したMLIPが構築され、学習データより大規模な系に対しても高精度で計算できること(転移可能性)が示された[5]。
従来のMLIPが限定的な元素種への対応に留まっていたのに対し、PFPは、45元素という高い汎用性を世界に先駆けて実現した[6]。この飛躍を可能にしたのが、独自のグラフニューラルネットワークアーキテクチャと、仮想的・不安定な構造までも意図的に組み込んだ学習データである。PFPは、従来のMLIPの常識を覆す、まさに画期的な存在である。著者が所属するMatlantis株式会社では、このPFPを利用可能な計算プラットフォーム「MATLANTIS™」を提供している[7]。PFPは約半年に一度のペースで改良が続けられており、2024年9月には自然界に存在する全ての安定元素(96元素)対応を達成した。ここまででPFPはDFTの精度を高速かつ広範囲で再現するuMLIPとなったが、さらに実験と計算の誤差を小さくできれば、より効率的な材料開発が期待できる。 一般にDFTは、高精度な交換相関汎関数を用いることで計算結果の精度を高められるが、計算時間が増大するというトレードオフを抱えている。一方、MLIPは一度学習が完了すれば、その推論時間は、学習に用いた計算データの精度(汎関数の種類など)には依存しない。つまり、より高精度な計算結果をMLIPに学習させることで、推論時間はこれまで同等のまま、より高精度な結果を得ることが可能になる。このアプローチを具現化したのが、2025年6月にリリースされたPFP v8である。PFP v8では、これまで用いていたPBE汎関数より高精度なr²SCAN汎関数による計算結果を学習させたr²SCAN modeを搭載し、実験値との誤差を改善できることを確認している。
PFPが切り開いたuMLIPというフロンティアは、2025年現在、学術界・産業界を巻き込んだ一大潮流となっている。多くの企業や研究機関が独自のuMLIPを開発し、分野全体が急速な発展を遂げている。この活況を受け、uMLIPを実践的に活用するための論文も出版された[8]。PFP開発者の一人である高本氏が本論文の著者に名を連ねていることは、PFPがこの分野を牽引する存在であることを示唆している。
4. uMLIPを用いた材料開発
ここまで、uMLIP が開発された経緯と現状について概観してきた。しかし、uMLIPの真価は、実際の材料開発で活用されてこそ発揮される。本項では、PFPを用いた具体的な材料開発事例を紹介する。
4.1 アンモニア合成触媒のスクリーニング
上述したハーバー・ボッシュ法の触媒開発は、uMLIPによって開発プロセスがどう変わり得るかを示す好例である。PFPを用いることで、多様な金属元素からなる約2000種の触媒表面上でのN吸着エネルギーとN-N遷移状態エネルギーの計算により、従来よりも高活性と予測される新規触媒の候補を短期間で複数見出されたことが報告されている[9]。この大規模スクリーニングは、DFTでは計算時間の観点で、従来のMLIPでは元素種の観点から難しかったが、PFPの高速性と汎用性によって初めて実現可能となった。このように、PFPは速度と汎用性の両面から、新規触媒の発見を加速させると期待される。
4.2 ポリマーの熱分解挙動の解析
ポリマーの耐熱性評価やケミカルリサイクル技術において、熱分解挙動の理解は重要である。我々は、PFPを用いてエポキシ樹脂の熱分解MDを実施した。その結果、特定の温度域で樹脂の分解が進行し、その分解メカニズムや主要な生成物が、実験結果や従来の研究報告とよく一致することが確認された[10]。従来、こうした反応の記述にはReaxFFのようなポテンシャルが用いられたが、煩雑なパラメータ調整が課題であった。パラメータ調整が不要なPFPを用いることで、多様なポリマーの熱分解特性を実験を介さず高速にスクリーニングでき、耐熱性に優れた新規ポリマーの効率的な設計につながる可能性が示された。
4.3 MOFの分子吸着性の評価
2025年現在、最も注目されている材料といえば、ノーベル化学賞を受賞した多孔性配位高分子(PCP)、あるいは、金属有機構造体(MOF)であろう。PCP/MOFは金属と有機配位子の組み合わせにより多様な構造を設計でき、分子吸着・貯蔵材料として注目されている。しかし、その広大な探索空間ゆえに、所望の機能を持つ構造を探索することは困難であった。ShimadaらはPFPを用いて、PCP/MOFのガス吸着特性が、活性化エネルギーによって説明できることを示した[11]。また、Bonakalaらは古典ポテンシャルとPFPを組み合わせ、12万8千以上の既知MOF構造から所望の機能を持つものを高速スクリーニングする手法を提案している[12]。これまで、PCP/MOFの多様な元素構成はMLIPの適用を困難にしていたが、PFPの汎用性がこの課題を解決する手段となった。
5. 結論と今後の展望
本稿では、材料開発が計算化学とAIの融合によっていかに劇的な変革を遂げつつあるかを概観した。その歴史は、「精度と計算時間、汎用性のトレードオフ」から始まった。この長年の課題を克服すべく登場したMLIPは、精度と速度の両立を達成し、さらにuMLIPへと進化することで汎用性を獲得した。今やuMLIPは、材料開発のゲームチェンジャーとして、その材料開発プロセスを根底から再定義し始めている。もちろん、uMLIPをもってしても計算可能な時間・空間スケールは、現実の材料で起こる現象に比べれば依然として小さい。マクロな特性を予測するためには、実験データや計算データを統合的に解析するマテリアルズ・インフォマティクス(MI)のアプローチが不可欠である。そして、uMLIPはMI分野においても重要な役割を担うと期待されている。uMLIPは、多様な原子環境を学習した基盤モデルとして機能し、特定の物性予測などの個別タスクに転移学習することで、少量データでも高精度な予測モデルを構築できることが報告されている[13,14,15]。つまり、uMLIPは単なる高速化ツールに留まらず、材料開発を支える基盤となりつつあるのだ。この新たな基盤の上で、未来の社会に不可欠な革新的材料が、これまで以上に迅速に生み出されていくであろう。
6. References
[1]人類の生存を支えるアンモニア合成② ハーバー・ポッシュ法- 化学工業の幕開け, https://www.jaci.or.jp/gscn/img/page_04/GCS_008-web_v2.pdf (2025年10月28日閲覧).
[2]T. B. Blank et. al., J. Chem. Phys. 1995, 103, 4129.
[3]S. Lorenz, A. Groß and M. Scheffler, Chem. Phys. Lett. 2004, 395, 210.
[4]J. Behler and M. Parrinello, Phys. Rev. Lett. 2007, 98, 146401.
[5]J. S. Smith, O. Isayev and A. E. Roitberg, Chem. Sci. 2017, 8, 3192.
[6]S. Takamoto et. al., Nat. Commun. 2022, 13, 2991.
[7]Matlantis (https://matlantis.com/), software as a service style material discovery tool.
[8]R. Jacobs et. al., Curr. Opin. Solid State Mater. Sci. 2025, 35, 101214.
[9]アンモニア合成触媒開発のための触媒スクリーニング, https://matlantis.com/ja/calculation/ammonia-synthesis-catalyst-screening/ (2025年10月28日閲覧)
[10]エポキシ分子の熱分解シミュレーション, https://matlantis.com/ja/calculation/thermal-decomposition-of-epoxy-molecules/ (2025年10月28日閲覧)
[11]T. Shimada et. al., Adv. Sci. 2024, 11, 2307417.
[12]S. Bonakala et. al., arXiv preprint arXiv:2509.06719, 2025.
[13]Z. Mao, W. Li, and J. Tan, npj Comput. Mater. 2024, 10, 265.
[14]S. Y. Kim, Y. J. Park, and J. Li, arXiv preprint arXiv:2506.18497, 2025.
[15]T. Shiota, K. Ishihara, and W. Mizukami, Digit. Discov. 2024, 3, 1714.
-




 URLを
URLを
コピーしました
新着記事
NEW
京大・福井謙一記念研究センターで学ぶ、研究を加速するAI材料シミュレーション──ENEOSと共に「最高のCO2吸着剤」設計に挑む

【解説】AIはなぜそう予測したのか? PFP descriptorsとShapley値で解き明かす原子レベルの解釈性

マテリアルズインフォマティクス 解説記事 計算化学

ゼロから書くSMILES記法
解説記事 計算化学

名古屋大学×Matlantis「最先端理工学実験」レポート AIシミュレーションが実験系学生の探究心に火をつけた4日間の集中講義
インタビュー 計算化学

機械学習ポテンシャル入門: 材料開発を加速するシミュレーション技術

機械学習ポテンシャル 解説記事