サイクリン依存性キナーゼ2阻害剤の高精度で高速な結合エネルギー評価
概要
創薬研究において、リード最適化段階でのタンパク質-リガンド結合親和性の正確な予測は、化合物選択の成否を左右します。しかし、量子化学計算による高精度な相互作用計算は1構造あたり数十時間を要し、大規模な化合物ライブラリへの適用は現実的ではありませんでした。本事例では、汎用原子レベルシミュレーター「Matlantis」に搭載されている機械学習ポテンシャルPFPを用いて、がん治療のターゲットとして知られるサイクリン依存性キナーゼ2(CDK2)と6種類の阻害剤の結合エネルギーを評価しました。その結果、フラグメント分子軌道法(FMO法)と同等以上の精度(R²=0.95)を維持しながら、約34,000倍の高速化を達成しました。
背景と課題
生体内におけるタンパク質とリガンド間の相互作用をより深く理解し、合理的なリード化合物の最適化や結合親和性の予測を行うために量子化学計算は広く用いられてきました。そして近年では、タンパク質の柔軟性や構造変化(ゆらぎ)を考慮したアプローチの重要性が認識され始めています。
例えば、Takabaらの研究(J. Comput. Chem. 2022)[1]では、MDシミュレーションで得られた構造アンサンブルに対し、フラグメント分子軌道法(FMO法)を用いて動的にエネルギーを平均化するDA-FMO法を提案しました。単一の結晶構造のみを用いた場合と比較して、DA-FMO法では計算結合エネルギーと実験値との相関が大幅に向上することが示されています。
しかし、FMO法などの量子化学計算は高精度なエネルギー評価が可能である反面、タンパク質、リガンド、溶媒を含む数万原子系のエネルギーを計算する場合、1つの構造あたり数十時間の計算時間を要することも珍しくありません。したがって、多数の構造に対してFMO計算を実行するには、膨大な計算資源と時間を要し、実用上の大きな障壁となっていました。
ターゲットシステム
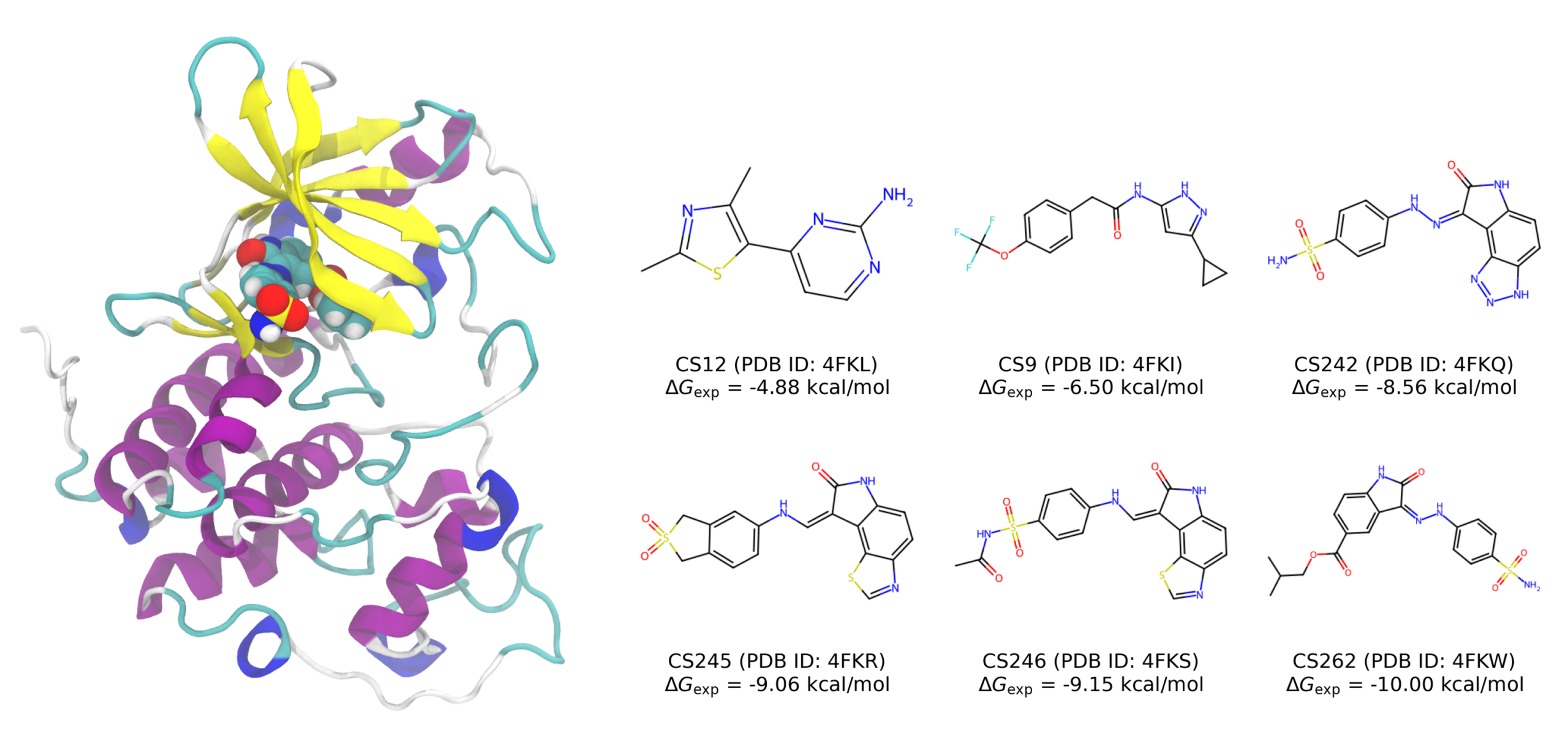
サイクリン依存性キナーゼ2(CDK2)はセリン/スレオニンキナーゼファミリーに属し、細胞周期の進行におけるフェーズ間の遷移を制御する重要なタンパク質です。CDK2の活性や制御の異常は腫瘍の成長と密接に関連しており、抗がん剤開発の有力なターゲットとして広く研究されています。
本事例では、結晶構造解析により結合様式が解明されている6種類のCDK2阻害剤(CS12, CS9, CS242, CS245, CS246, CS262)を計算対象としました。これらの阻害剤はいずれも生理的条件下で電気的に中性であり、実験結合自由エネルギー(ΔGexp)は−4.88〜−10.00 kcal/molの範囲にわたります。
計算手法
構造アンサンブルの準備
Takabaらが実施しFMODBに登録されているMDシミュレーションのトラジェクトリーデータを利用しました[1, 2]。系にはタンパク質、リガンド、溶媒を含めて約1万原子が含まれています。各タンパク質-リガンド構造について、50 ns×5のシミュレーションから12 ns以降のスナップショットを2 nsごとに抜き出し、合計100構造を結合エネルギー計算に利用しています。
結合エネルギーの算出
以下のような超分子的なアプローチによって、複合体、タンパク質、およびリガンドのエネルギー差からリガンド結合エネルギーを算出しました。
結合エネルギーは各リガンドにつき100スナップショット(合計600構造)について計算して、最終的にはそれらの値の平均と分散を計算しました。
比較対象の計算手法
表1に示すようにFMOとPFPで結合エネルギーを計算し、それぞれ実験結合自由エネルギー(ΔGexp)との相関を確認しました。
表1: 比較手法
| FMO (DA-FMO) | Matlantis (PFP) | |
| 計算レベル | FMO-MP2/6-31G* | PFPv8 (R2SCAN + D3) |
| エネルギー評価 | フラグメント間相互作用エネルギー (IFIE) | 超分子法による結合エネルギー(ΔEbind) |
| ソフトウェア | ABINIT-MP | Matlantis |
表2: 計算原子数
| タンパク質 | リガンド | 水分子 | 合計 | |
| CS12 (PDB: 4FKL) | 4848 | 24 | 6897 ~ 7524 | 11769 ~ 12396 |
| CS9 (PDB: 4FKI) | 4848 | 37 | 6861 ~ 7452 | 11746 ~ 12337 |
| CS242 (PDB: 4FKQ) | 4848 | 36 | 6963 ~ 7482 | 11847 ~ 12366 |
| CS245 (PDB: 4FKR) | 4848 | 39 | 6873 ~ 7533 | 11760 ~ 12420 |
| CS246 (PDB:4FKS) | 4848 | 42 | 6882 ~ 7479 | 11772 ~ 12369 |
| CS246 (PDB:4FKS) | 4848 | 49 | 6879 ~ 7467 | 11776 ~ 12364 |
結果
結合エネルギーの予測精度(ΔEbind vs ΔGexp)
各阻害剤について、計算により得られた結合エネルギー(ΔEbind)と実験結合自由エネルギー(ΔGexp)との相関を評価しました。FMO法(MP2/6-31G*)およびPFPv8(R2SCAN + D3)で結合エネルギーを評価しました。結果は表3および図2のとおりです。
表3: 予測精度の比較
| 手法 | R² | RMSE (kcal/mol) |
| FMO (DA-FMO) | 0.91 | 0.52 |
| PFPv8 (R2SCAN + D3) | 0.95 | 0.38 |
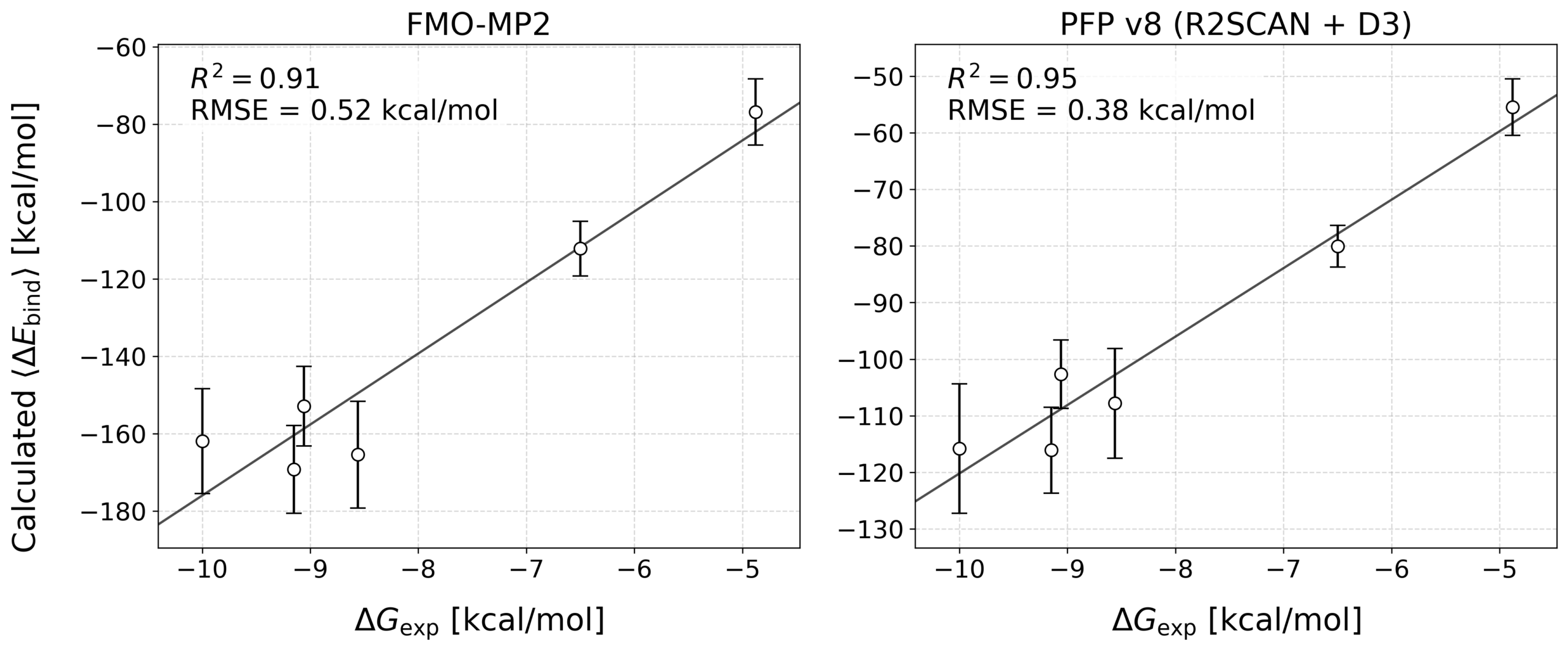
PFPはFMOと比較して誤差(RMSE)が小さく、実験の結合自由エネルギーに対して非常に良好な相関を示しました。すなわち、従来の量子化学計算に匹敵する高い精度でタンパク質-リガンド間の相互作用を評価できることが確認されました。
計算速度の比較
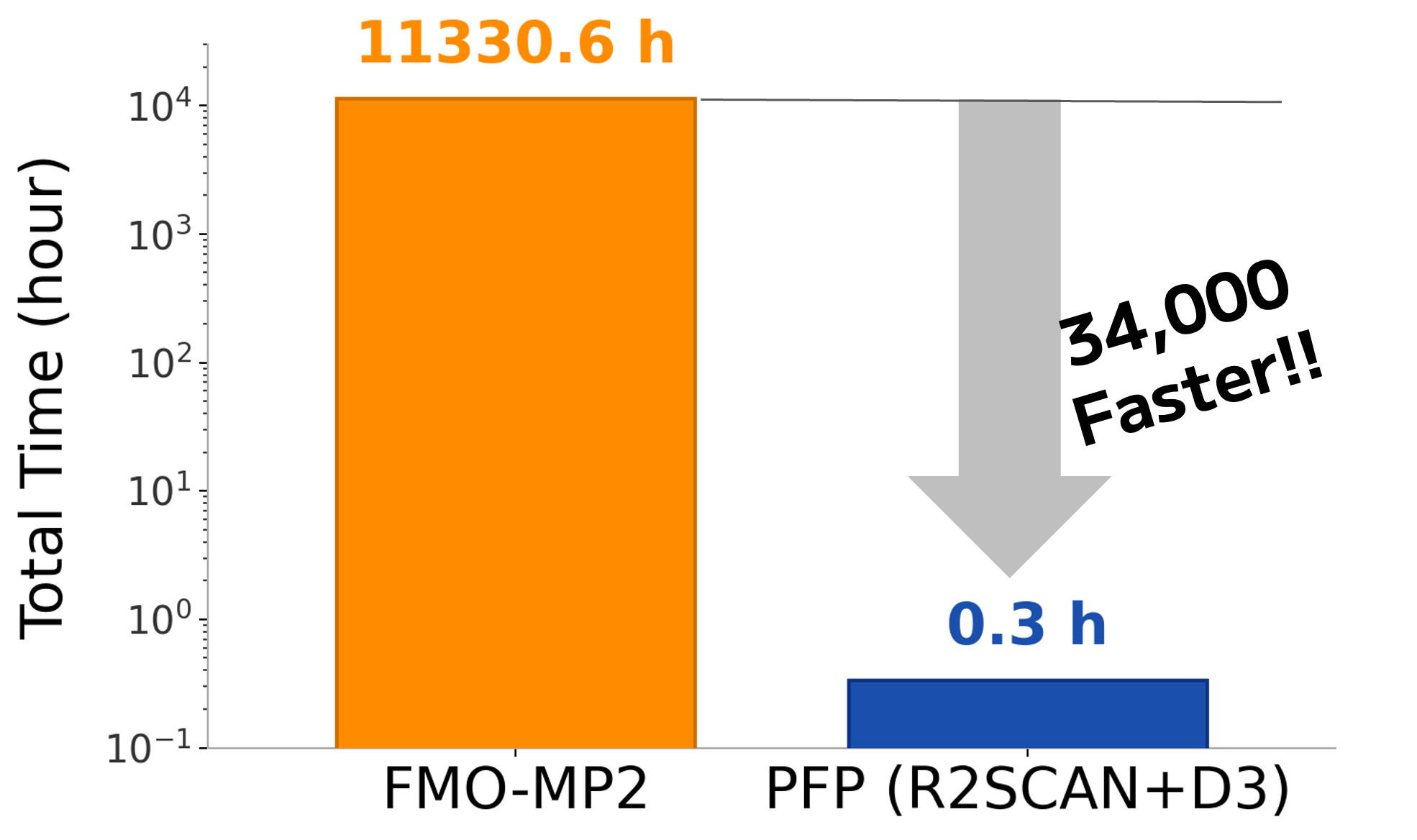
続いて実行に要した計算速度について比較しました。合計600構造に対する計算において、FMOは計11,330時間(計472日分)の計算時間を要したのに対し(※)、Matlantis(PFP)はわずか20分で計算を完了しました。すなわち、FMOと比較して約34,000倍高速に結合エネルギー計算を実行することができました。
この速度差をスクリーニング規模に換算すると、1,000化合物×100スナップショット(計100,000構造)の評価にFMOでは約5.2年を要するのに対し、Matlantisでは約56時間で完了できる計算になります。前述のとおり予測精度はFMOと同等以上であったことからも、これまで計算コストの制約で断念せざるを得なかった大規模な化合物探索にも、量子化学計算レベルの相互作用評価を適用できることが示されました。
※FMOの計算時間はFMODB[1, 2]から取得したログファイルに基づき算出しています。
まとめ
MDシミュレーションから得られたアンサンブル構造に対する相互作用エネルギーの平均化は、結合親和性の予測精度向上に極めて有効です。しかし、従来の量子化学計算をスクリーニング用途に適用するには、膨大な計算資源と時間を要することが大きな障壁でした。
本事例では、Matlantis(PFP)を活用することで、量子化学計算レベルの高精度な予測を維持したまま、計算時間を大幅に短縮できることを示しました。これまで計算コストの壁により困難であった大規模な化合物空間に対しても、量子化学計算と同等精度の相互作用評価を効率的に行うことが可能となります。これにより、創薬や農薬設計において有望な化合物探索を大幅に加速することが期待されます。
参考文献
[2] FMO Database (FMODB): https://drugdesign.riken.jp/FMODB/
タグ
本事例の公開日:2026.05.20
-




 URLを
URLを
コピーしました