テーマ概要(背景)
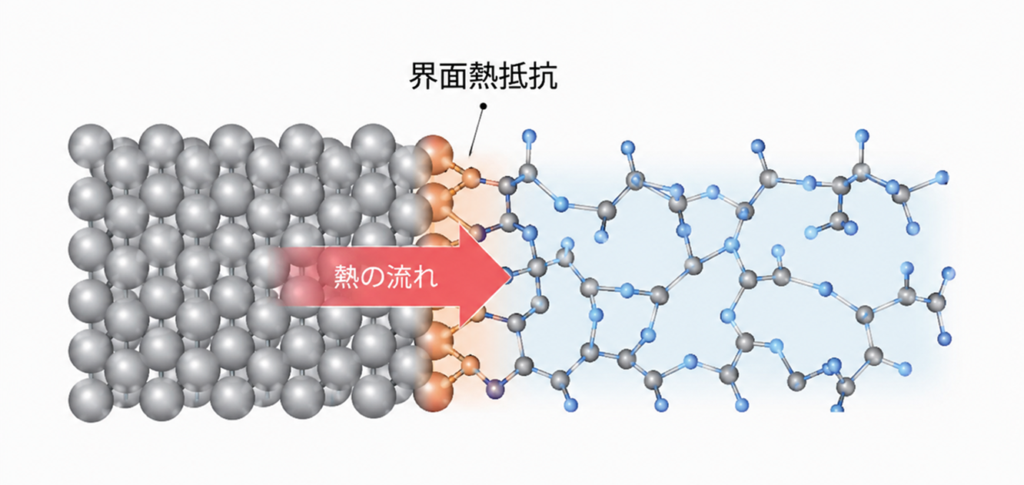
界面熱抵抗は、半導体、パワーエレクトロニクス、二次電池など様々な分野で重要となる物性です。金属-金属界面、金属-ポリマー界面など材料種の組み合わせは多岐にわたりますが、特に近年は、半導体産業の後工程(パッケージング)におけるサーマルマネジメントの重要性が高まる中で、基板上のポリマーの熱物性を正確に把握する観点から関心を集めています。
従来、界面熱抵抗の評価には古典力場が用いられてきましたが、異種材料界面における複雑な化学結合を正確に記述することが困難であり、予測精度には限界がありました。Matlantisを用いると、元素種を問わず、界面の接着性を考慮したうえで計算を実行できます。本事例では、2種類の金属基板(Au, Ni)とPMMAの接着界面のモデリング方法と、界面熱抵抗の検証結果を報告します。
計算モデルと計算方法

本計算では、接着構造のモデリングを高速化するために、分子シミュレーションソフトウェアOpenMMに実装されている古典力場とASECalculatorの連携機能を用いました。以下の手順に従い、金属のスラブモデルとポリマー(PMMA, 5量体)の接合モデルを作成しました。
- ポリマーの目標密度の80%に到達するまで、古典力場でMD計算を実行しました(ポリマーはGAFF2力場、金属原子は固定し、ポリマー・金属間はLJポテンシャルを適用)。
- 界面での接着性を高精度に計算するため、残り20%の圧縮はPFP/MM手法に切り替えました。PFP領域は界面近傍、MM領域はそれ以外の領域です。
PFP/MM境界ではPFPの視点では外側が真空に見えるため、2つの工夫を入れました。ポリマー側は境界が共有結合を切断するため、Link Atom法で切断結合の先に仮想水素を付加し、PFPに妥当な結合環境を見せます。この原子はMDの更新には利用されません。金属側はPFP領域の外側にPFPには見えるが動かさないバッファ層を設け、境界の原子がバルク金属を見て自然な力が加わるようにしました。古典計算の段階ではポリマー・金属間に化学結合は形成されませんが、PFP/MM に切り替わると両者が接着します(図1参照)。
計算は、Matlantis上で試験的に実装されているGPUノートブックインスタンスで実行しました。これにより、古典力場の計算をGPUで高速化し、時間のかかるPFP領域の計算をサイズ削減により短縮できるため、PFP単独では数日要することもあった圧縮計算を30分程度で完了することができました。この手法を用いて、3000原子程度のAu, Ni/PMMAモデルおよび断面積の大きい7000原子程度のAu,Ni/PMMAモデルの計4通りのモデルを高速に作成しました。
計算結果と展望 ①
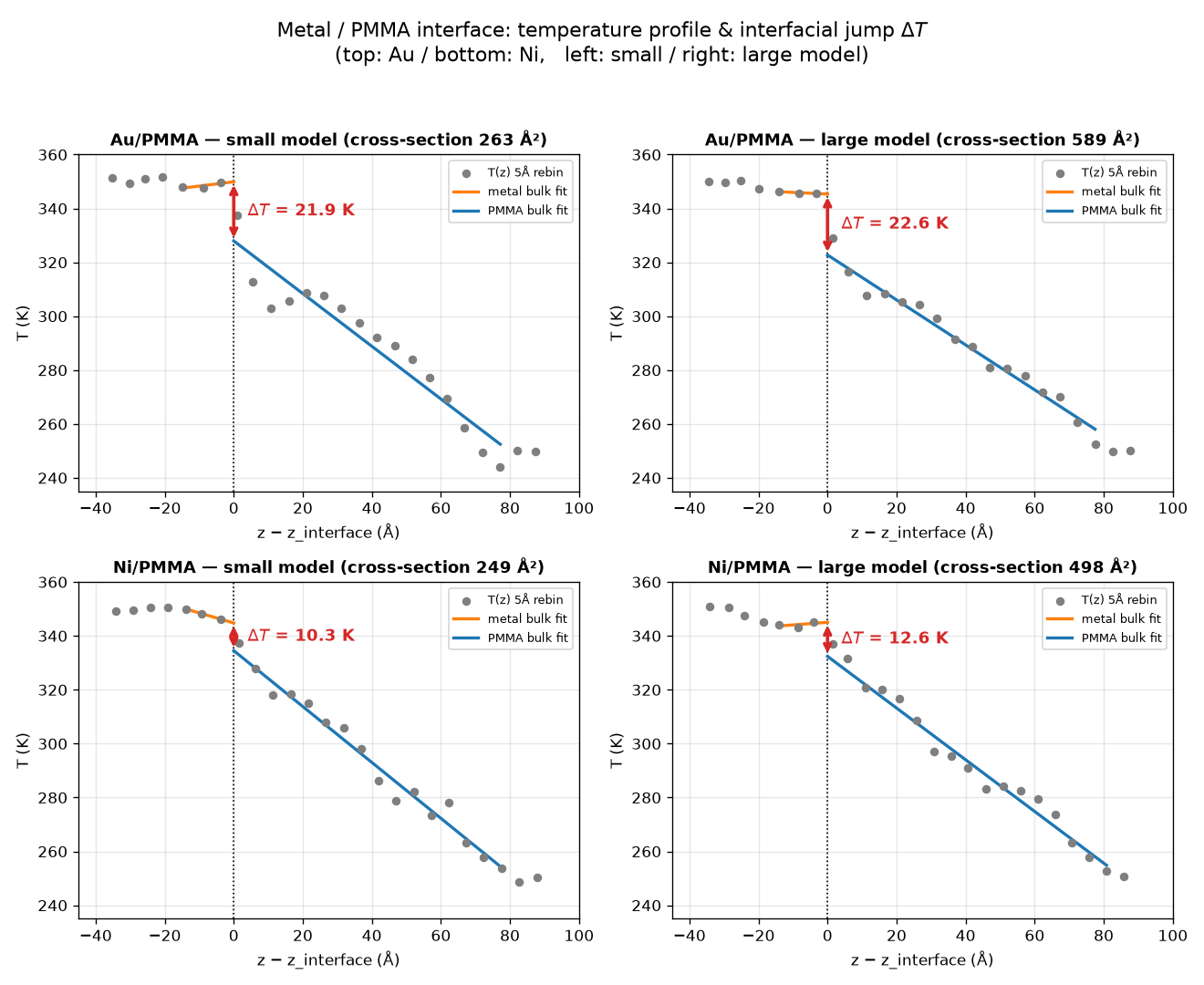
得られた接着構造に対し、Matlantis-LAMMPSインターフェースを用いて、構造最適化→NPT→NVT計算による平衡化処理を施した後、金属側に高温熱浴(350K)、ポリマー側に低温熱浴(250K)を設定して500psの非平衡分子動力学(NEMD)計算を実行しました。NEMD計算では全原子にPFPを割り当てました。得られたスナップショットに対し、系を熱流方向に沿って多数のスラブに区切り、各スラブ内の運動エネルギーから定義した平均温度を断面温度としました。熱浴領域を除いた金属側・ポリマー側それぞれのバルク熱伝導領域を線形フィットし、界面位置へ外挿した不連続分を界面温度ジャンプΔTと定義しました。結果が図2です。この結果から以下が読み取れます:
–Niに比べAuの温度ジャンプは大きい(Au ~ 22Kに対しNi ~ 10-13K)。これはPMMAと金属の接合性の違いを示唆しています。実際、Auは主に分散力相互作用で吸着しており、界面の熱輸送が制限されることがこの差に反映されていると解釈できます。
–小型モデルと大型モデルでほぼ同等の温度ジャンプが得られました(Au:21.9 vs 22.6K、Ni:10.3 vs 12.6K)。界面物性が横断面サイズに対して収束していることを意味し、計算時間を節約したい場合は小型モデルでも妥当な推定に活用できることを示唆します。
計算結果と展望 ②
また、4モデルにおける界面熱抵抗(カピッツァ抵抗)を以下の物理式に基づいて算出しました。
ここでJは界面を通過する熱流[W]、Aは界面断面積[m2]です。結果、各モデルの熱抵抗値は表1のようになりました。Au/PMMAの界面熱抵抗の実験値は16.9×10-9 m2K/Wと報告されており、計算値は実験値の約1/2となっています[1]。実験の界面には汚染や粗さが含まれるのに対し、本計算は清浄な理想界面を扱っているため、実験より熱を通しやすい(熱抵抗が小さい)結果になるのは自然な帰結です。また同じ文献[1]では熱伝導性を高めるためにAuとPMMAの間に厚さ2nmのNi層を挿入すると界面熱コンダクタンス(界面熱抵抗の逆数)が素のAu/PMMA界面の2.4倍に向上すると報告されています。本計算でもNiの熱抵抗値は相対的に低くなっており、この結果とも整合しています。なお、Niは界面抵抗が小さくΔTが小さいため、Auに比べ算出値の不確かさは大きめです。
以上、OpenMMによる高速な界面モデリングと界面熱抵抗算出の結果をお示ししました。界面物性は様々な産業領域で課題となる重要な物性の1つです。Matlantisは現実的な界面をチューニングなしで扱える優れた特性を備えています。このモデリング方法は界面熱抵抗計算だけではなく、界面の引張シミュレーションなどにも活用でき、様々な応用が生まれることを期待しています。
表1. Au/PMMAとNi/PMMAの界面熱抵抗
| モデル(金属/ポリマー) | 界面熱抵抗 RK [m² K W⁻¹] |
| Au/PMMA (small) | 7.9 × 10⁻⁹ |
| Au/PMMA (large) | 7.5 × 10⁻⁹ |
| Ni/PMMA (small) | ~ 2 × 10⁻⁹ |
| Ni/PMMA (large) | ~ 5 × 10⁻⁹ |
計算条件
| 計算条件 | 詳細 |
| PFP model version | v9.0.0 |
| calc_mode | r2scan_plus_d3 |
| 系のサイズ | Au/PMMA:3,497原子、7,754原子 Ni/PMMA:3,703原子、7,406原子 |
| 計算時間 | モデリング:30分程度(Matlantis GPUノートブックを利用) NEMD計算:小規模モデル 2日程度、大規模モデル 4日程度 |
| MD計算条件 | シミュレーション時間 = 500 ps, timestep = 1 fs |
| 温度設定 | 高温熱浴(金属側):350 K, 低温熱浴(ポリマー側):250 K |
| アンサンブル | 平衡化:構造最適化→NPT→NVT(300K) 本計算(NEMD):NVE + 熱浴領域のみLangevinサーモスタット |
参考文献
[1] S. Sandell et al., Nanomaterials 10, 670 (2020). https://doi.org/10.3390/nano10040670
-




 URLを
URLを
コピーしました