User Testimonials




Simulation Case Studies
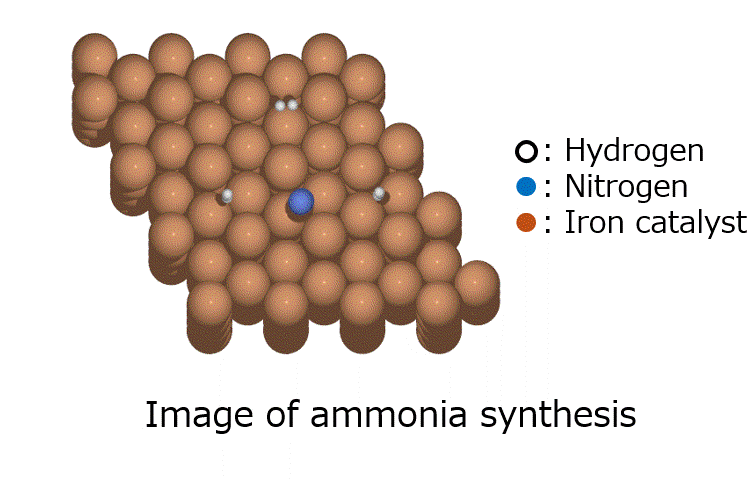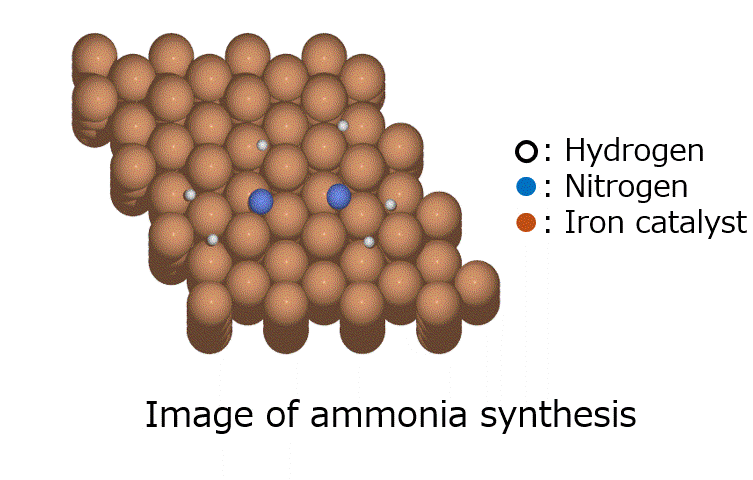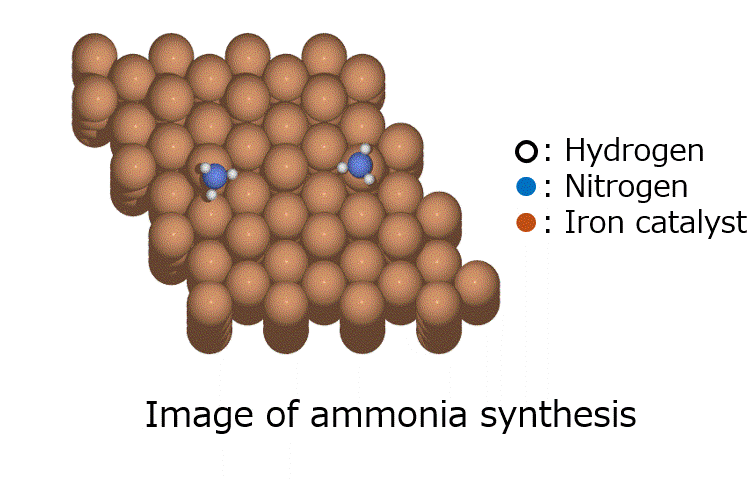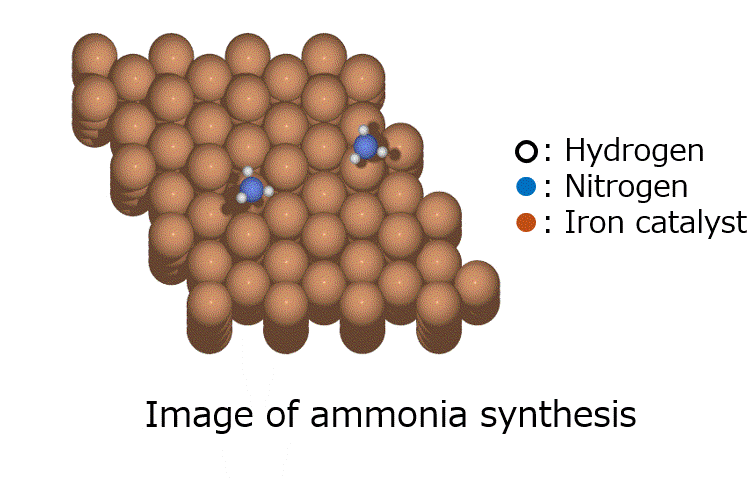
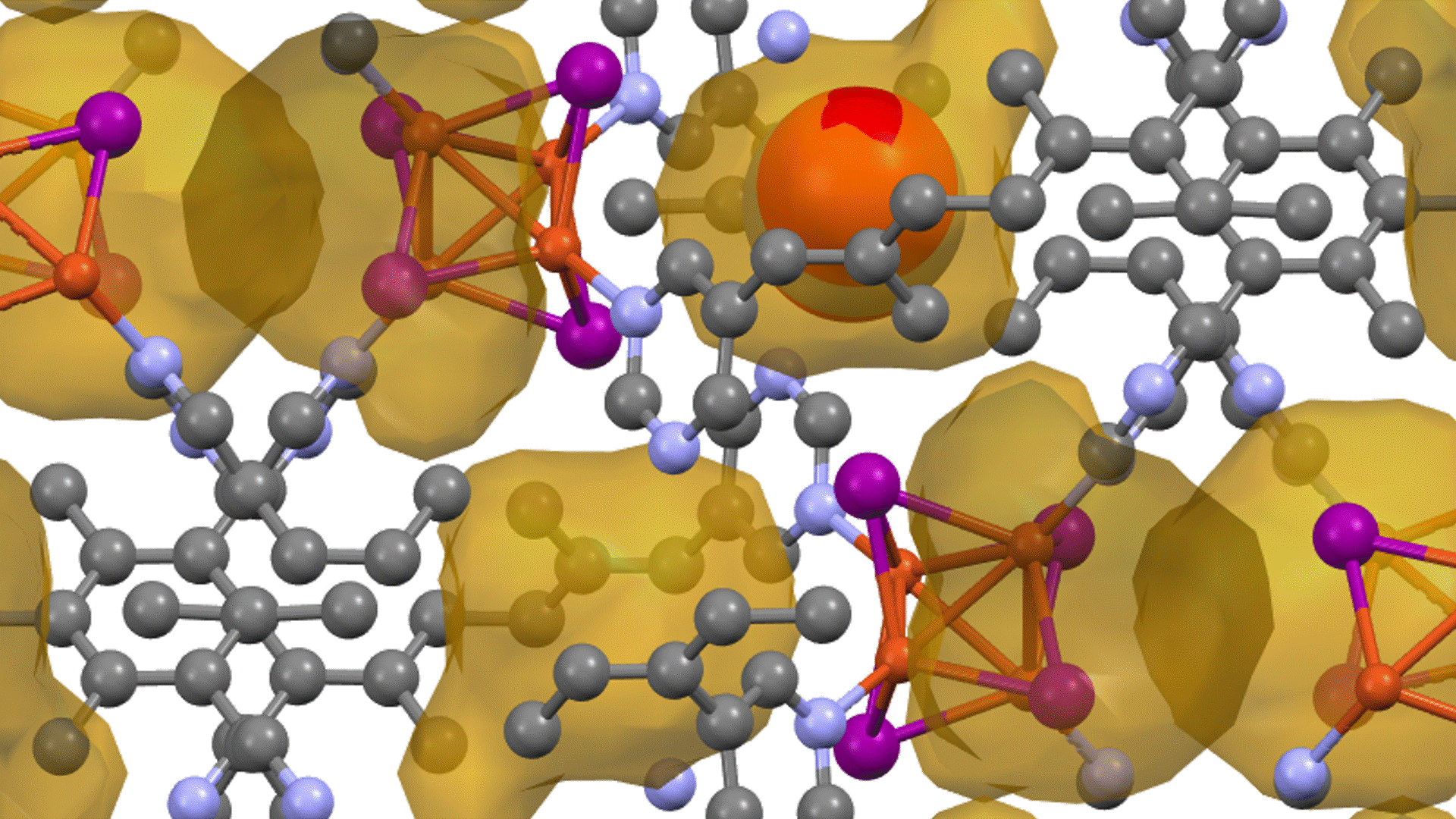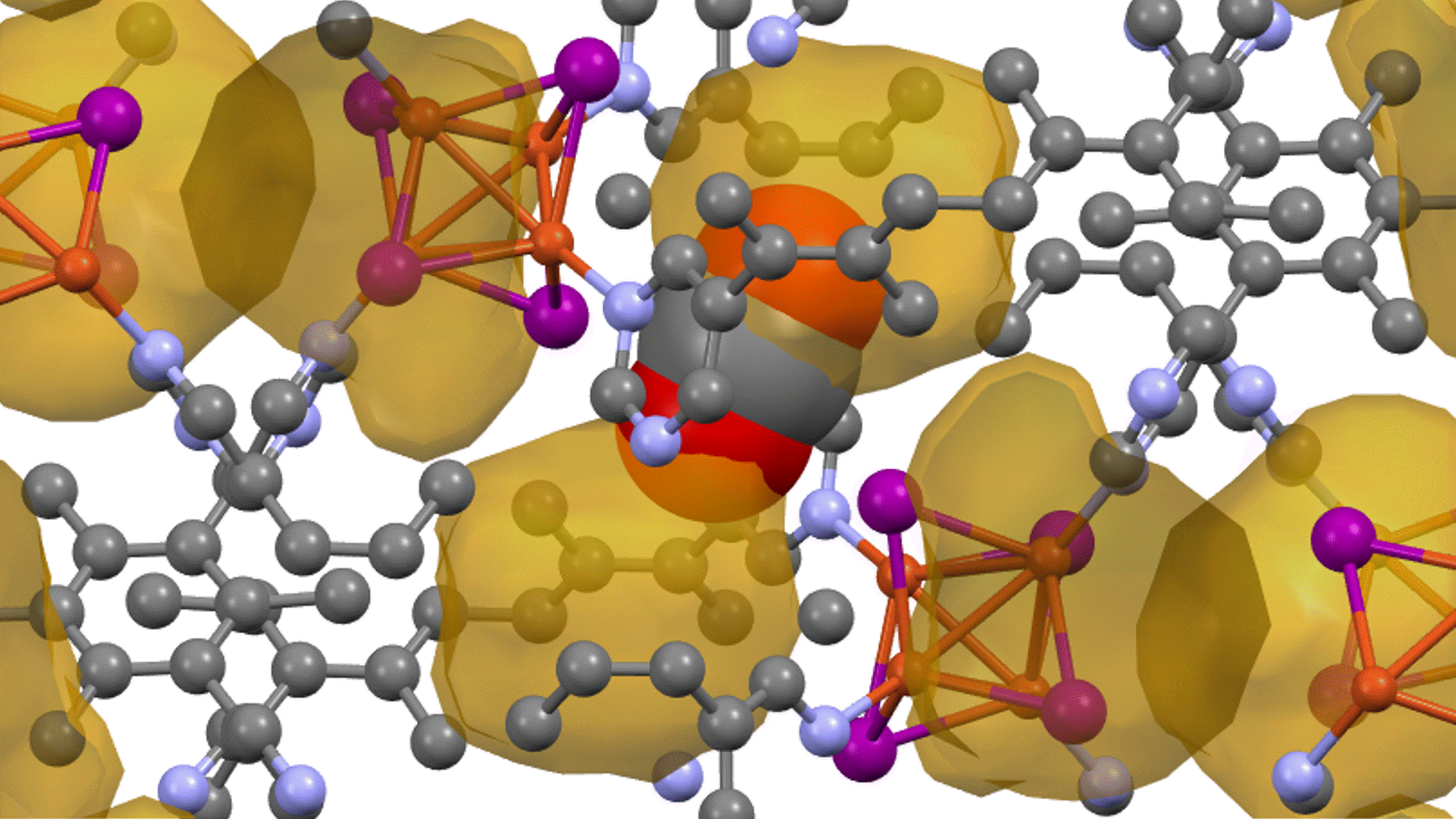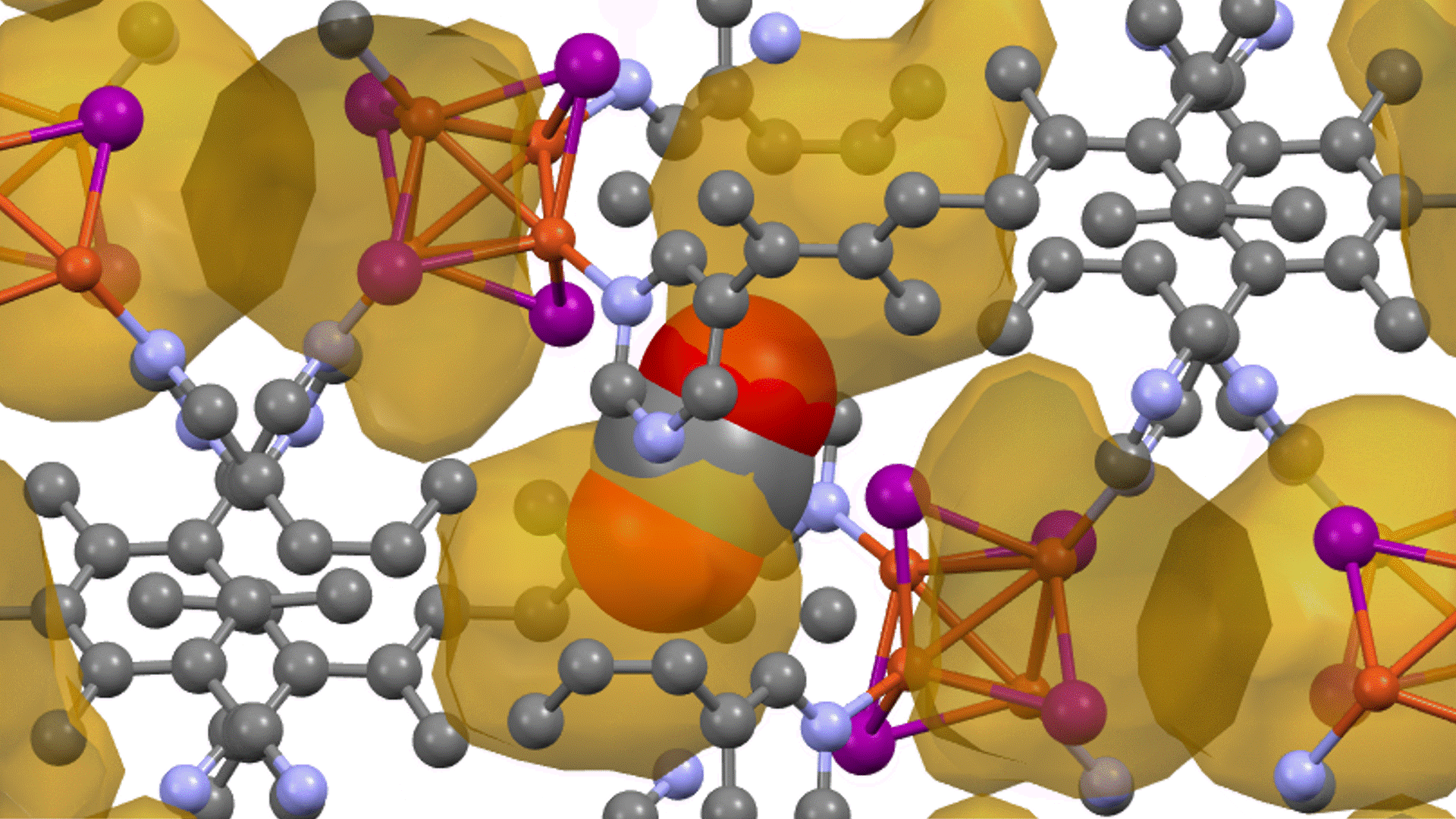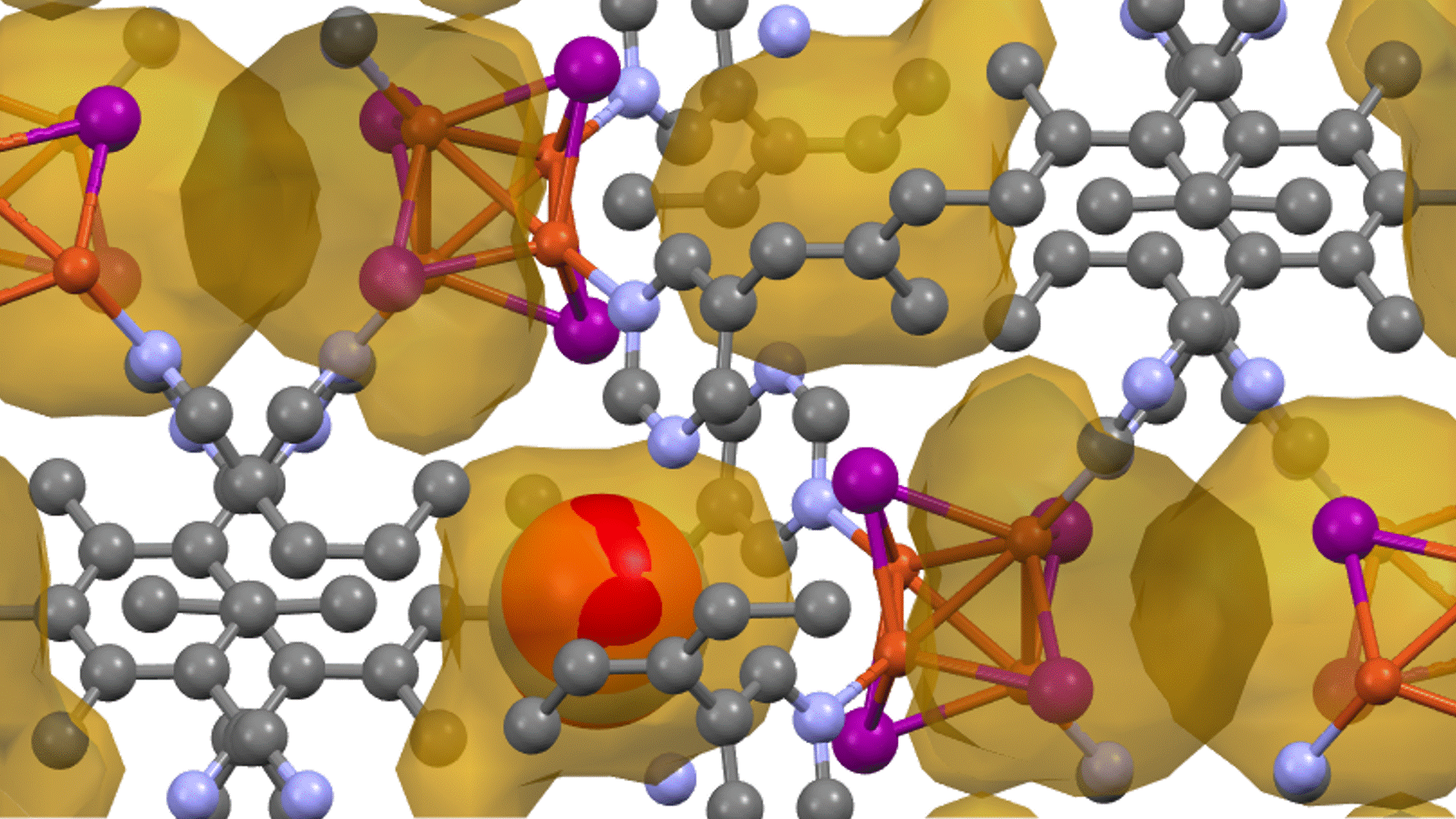
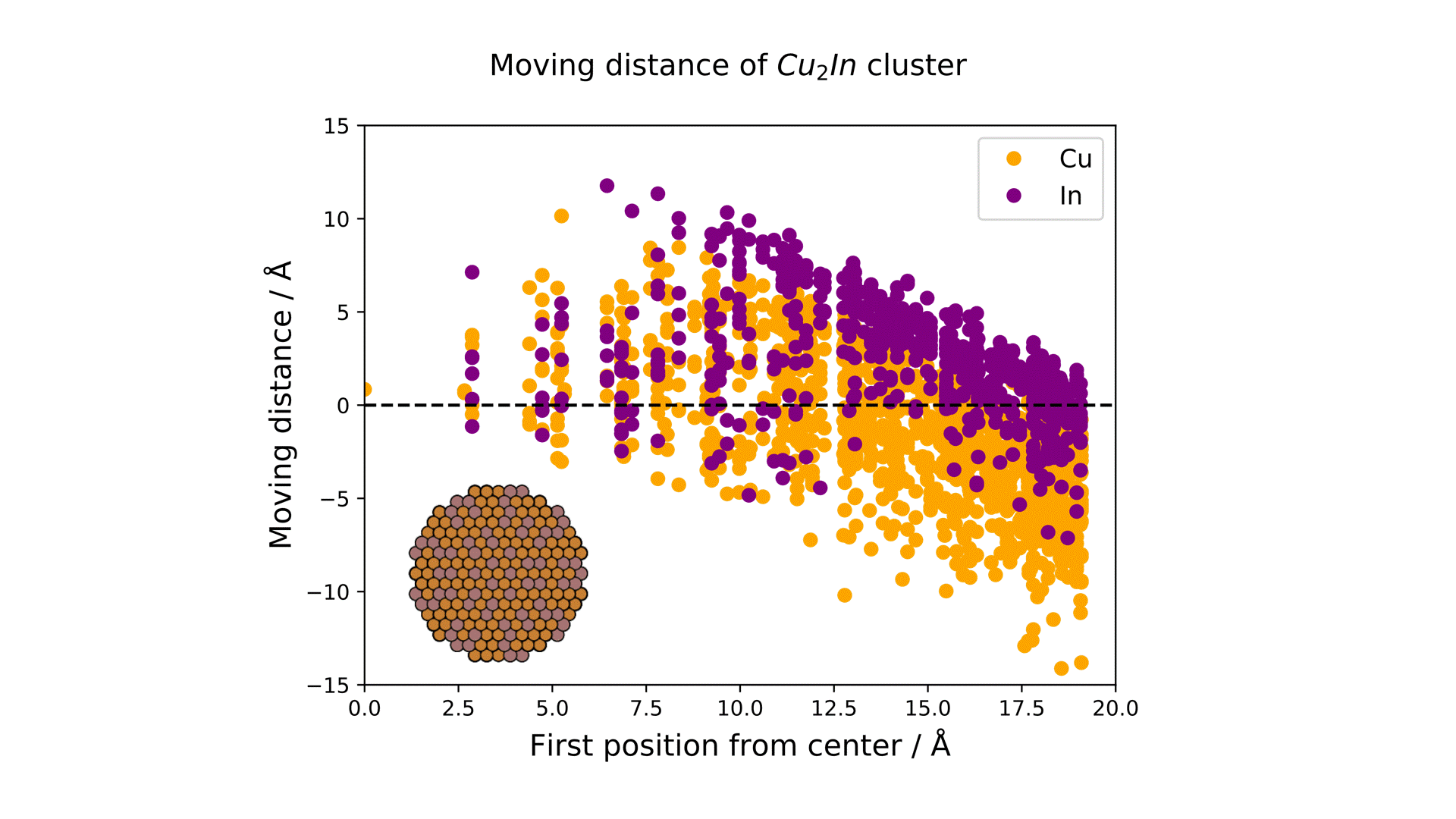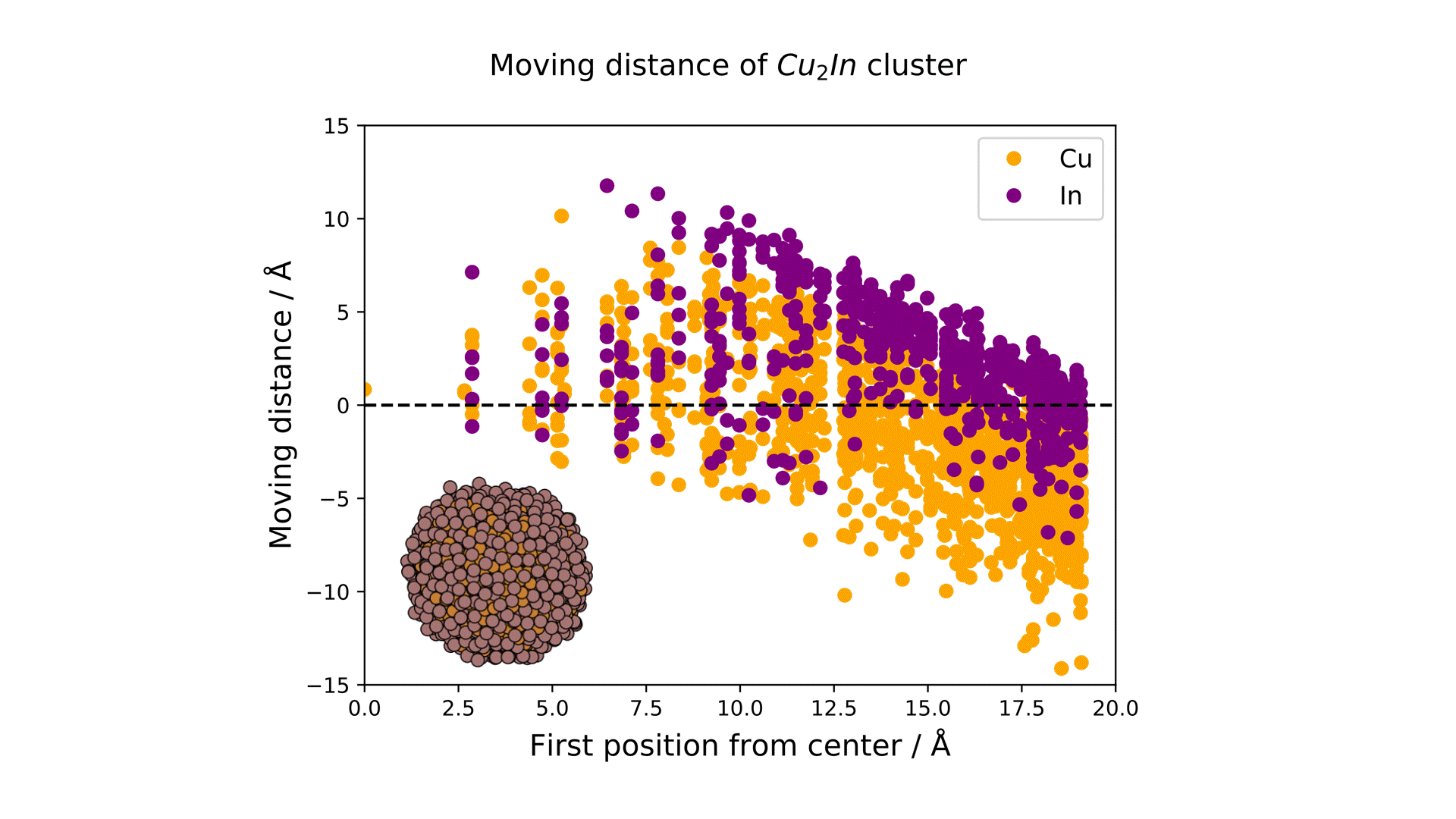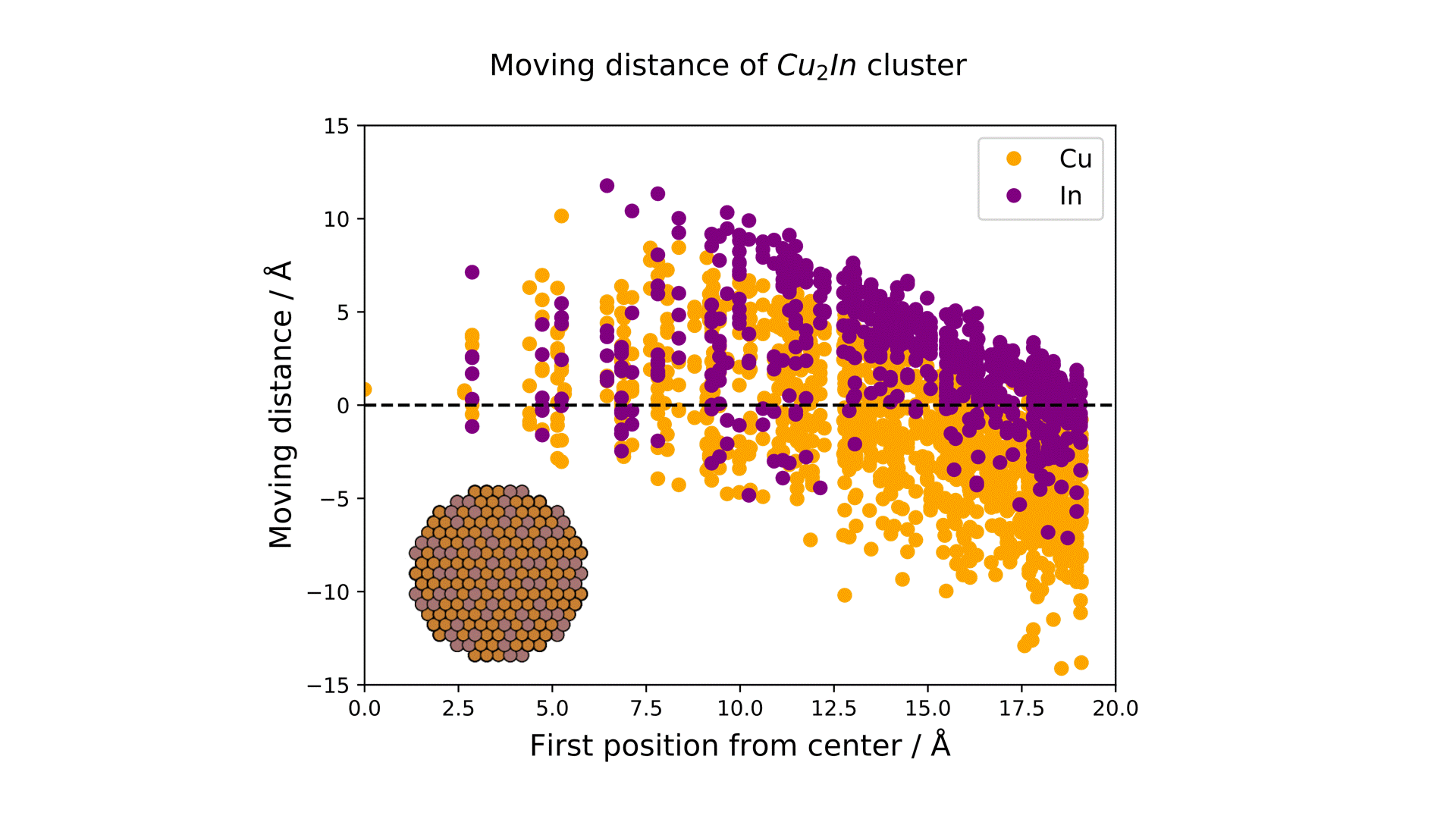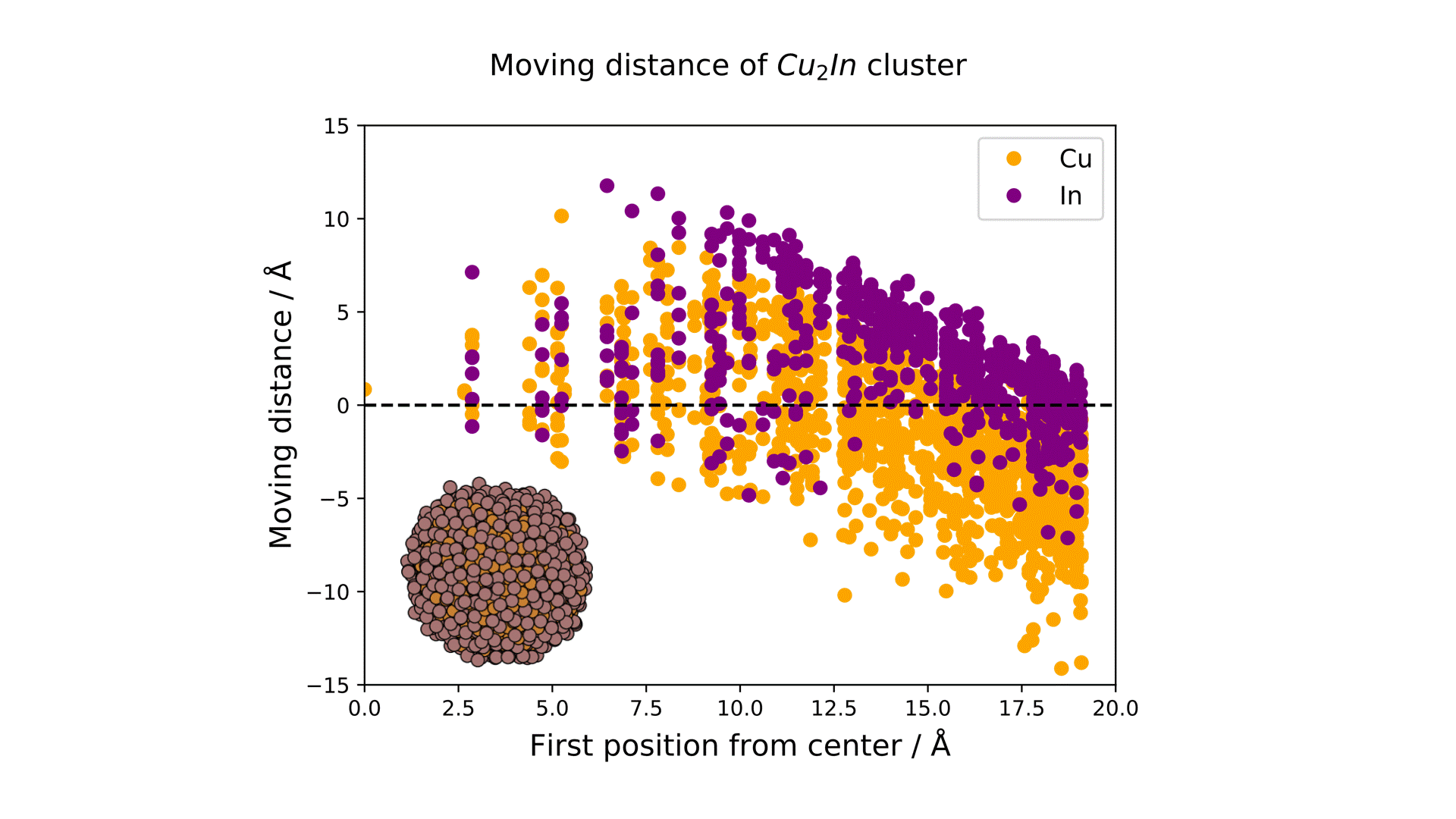
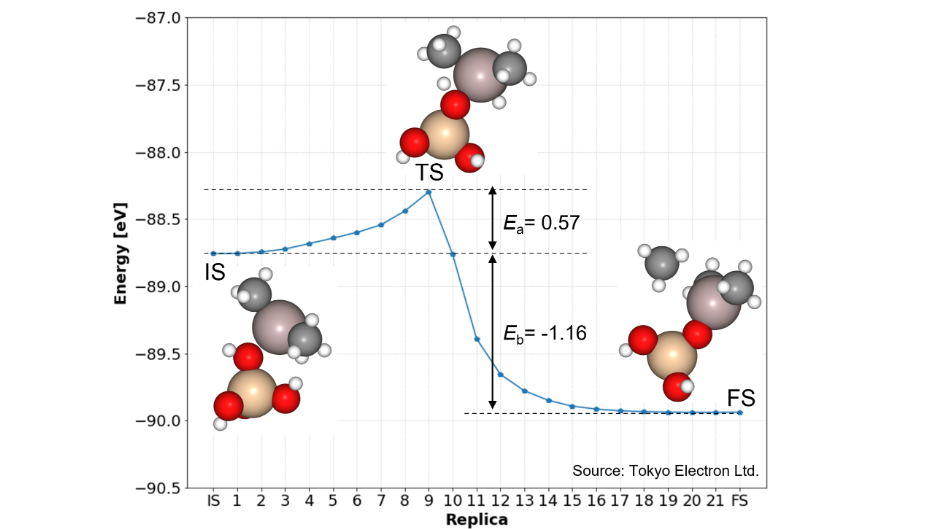
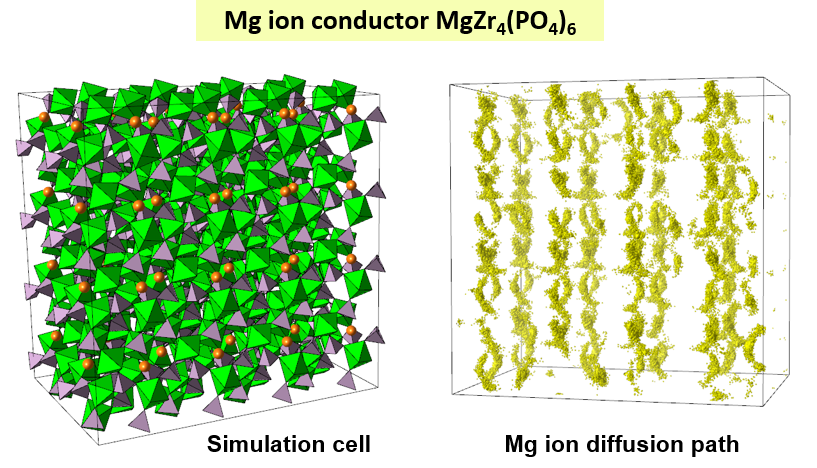
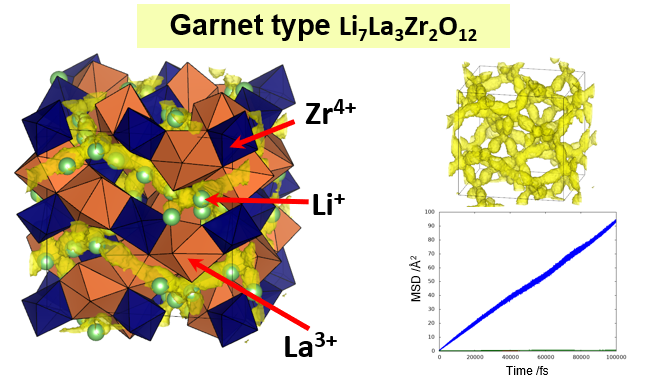
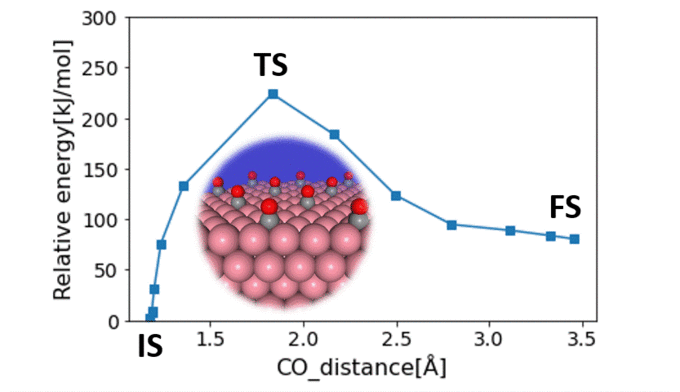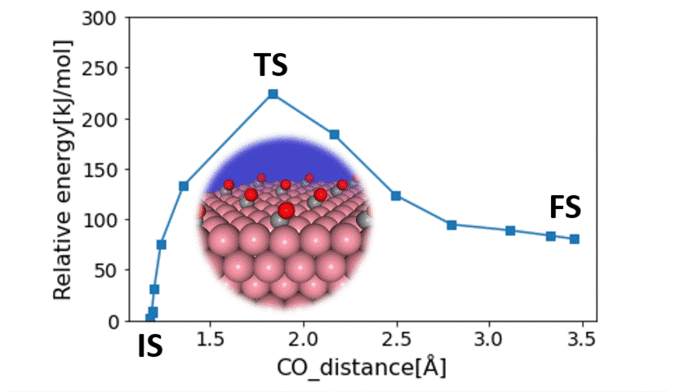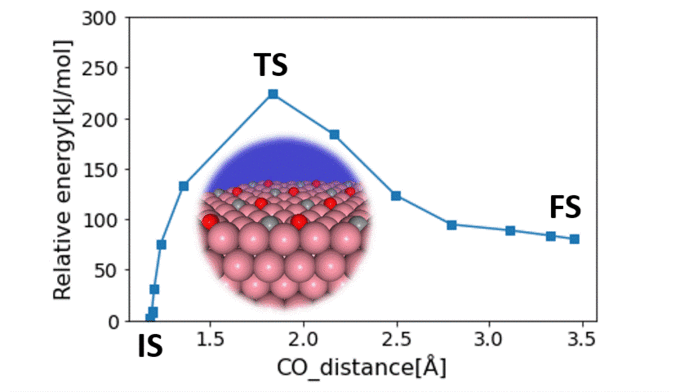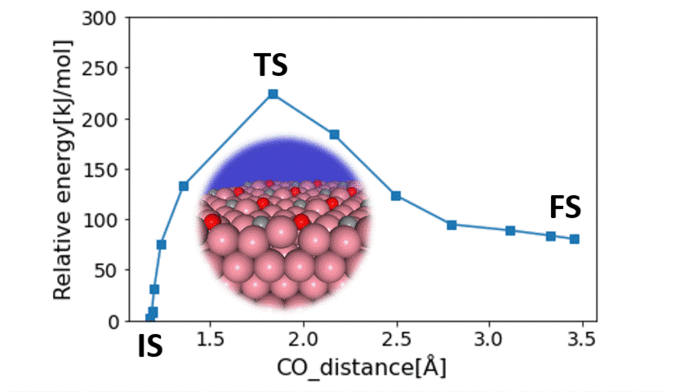
Research Using Matlantis
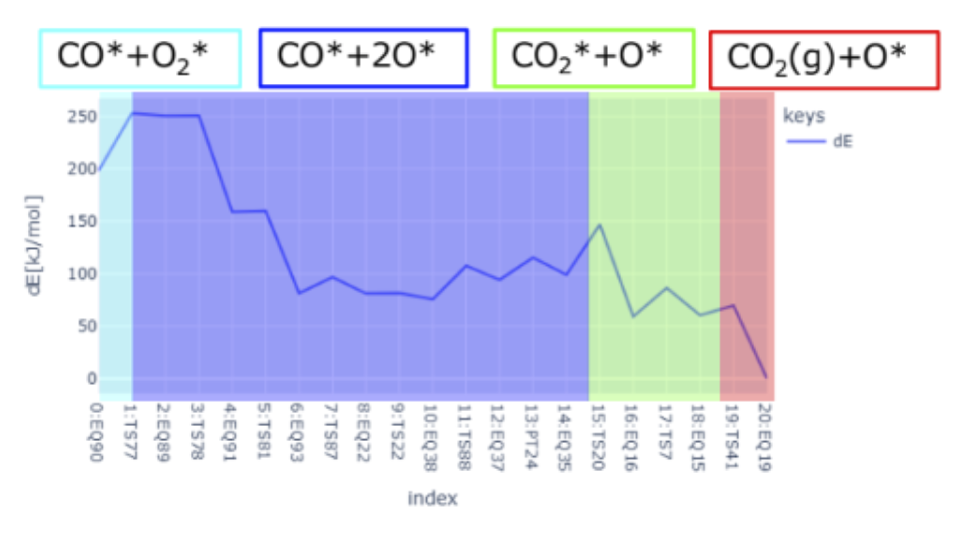
Surface Steric Effect in Heterogeneous Catalysis as the Origin of the High Activity Induced by Strong Metal–Support Interactions
Gerardo Valadez Huerta, Kaoru Hisama, Katsutoshi Sato, Katsutoshi Nagaoka, Michihisa KoyamaSupported nanoparticles offer unique opportunities for enhancing catalytic activity via strong metal–support interaction (SMSI). Even with state-of-the-art experimental techniques, the atomistic origin of this enhancement remains unclear, while current computational limitations make it difficult to provide a theoretical explanation. This study focused on clarifying the atomistic mechanism of SMSI by investigating N2 dissociation from Ru/La0.5Ce0.5O1.75-x catalysts. Fast calculations using a neural network potential enabled the analysis of 328 complex nanoparticle models with varying degrees of site heterogeneity, encompassing over 25,768 adsorption sites. Our findings were validated against infrared spectra and helped identify catalyst configurations with enhanced catalytic activity, driven by SMSI. Specifically, the dissociation path of N2 molecules sandwiched between decoration cations on a nanoparticle near the support exhibited a low activation barrier. Our theoretical approach represents a major advancement in bridging the gap between simulation and empirical data and in our understanding of complex supported nanoparticle catalysts.
Read moreAnion Intercalation and Selectivity in NiFe LDH: Insights from Neural Network-Enhanced Molecular Dynamics
Tien Quang NGUYEN, Susan Meñez ASPERA, Yingjie CHEN, Kaoru HISAMA, Michihisa KOYAMAMolecular dynamics (MD) simulations, accelerated by a universal neural network potential, were employed to investigate the dynamic behaviors of ten inorganic anions (Br-, Cl-, F-, OH-, NO3-, H2PO2-, H2PO3-, HPO42-, CO32- and SO42-) intercalated into NiFe layered double hydroxide at varying hydration levels. Our results show that the lattice parameters along the layered double hydroxide (LDH) layers are minimally affected by the intercalated anions and water content, while the lattice parameter perpendicular to the layers, i.e., the basal spacing, is strongly influenced by the type of anion and hydration level. The basal spacing is closely correlated with the ionic radius and charge of the anions, as well as the amount of water uptake. Mean squared displacement (MSD) analysis reveals distinct behaviors of the anions under different hydration conditions. While some anions, such as NO3- and H2PO2-, exhibit noticeable mobility, others remain largely immobilized, primarily due to strong electrostatic interactions with the LDH layers and water molecules. Hydration weakens the interaction between anions and the LDH but also restricts the mobility of anions due to the formation of hydration shells. These findings provide insights into anion dynamics and selectivity in NiFe LDH, which are critical for designing materials for anion removal applications.
Read moreLong Time CO2 Storage Under Ambient Conditions in Isolated Voids of a Porous Coordination Network Facilitated by the “Magic Door” Mechanism
Terumasa Shimada, Pavel M. Usov, Yuki Wada, Hiroyoshi Ohtsu, Taku Watanabe, Kiyohiro Adachi, Daisuke Hashizume, Takaya Matsumoto, Masaki KawanoA coordination network containing isolated pores without interconnecting channels is prepared from a tetrahedral ligand and copper(I) iodide. Despite the lack of accessibility, CO2 is selectively adsorbed into these pores at 298 K and then retained for more than one week while exposed to the atmosphere. The CO2 adsorption energy and diffusion mechanism throughout the network are simulated using Matlantis, which helps to rationalize the experimental results. CO2 enters the isolated voids through transient channels, termed “magic doors”, which can momentarily appear within the structure. Once inside the voids, CO2 remains locked in limiting its escape. This mechanism is facilitated by the flexibility of organic ligands and the pivot motion of cluster units. In situ powder X-ray diffraction revealed that the crystal structure change is negligible before and after CO2 capture, unlike gate-opening coordination networks. The uncovered CO2 sorption and retention ability paves the way for the design of sorbents based on isolated voids.
Read moreOxidative Dehydrogenation of Ethane Combined with CO₂ Splitting via Chemical Looping on In₂O₃ Modified with Ni–Cu Alloy
Kosuke Watanabe, Takuma Higo, Koki Saegusa, Sakura Matsumoto, Hiroshi Sampei, Yuki Isono, Akira Shimojuku, Hideki Furusawa and Yasushi SekineModified In2O3 has the potential to be a better oxygen storage material due to its readily reducible surface and abundant bulk lattice oxygen released with a marked valence change from In3+ to In0. This work describes that In2O3 modified with a Ni–Cu alloy supports a chemical looping system consisting of oxidative dehydrogenation of ethane and CO2 splitting at the low temperature of 873 K with a large oxygen capacity (>4 wt %). This reaction system is achieved through dynamic changes between Ni–Cu binary alloy and Ni–Cu–In ternary alloy associated with the redox of indium species. Meticulous material screening, characterization, and theoretical calculations have revealed that the Ni–Cu alloy promotes the redox of In2O3 by activating ethane and by incorporating reduced indium species.
Read moreMolecular Dynamics Simulation of Polymer Electrolyte Membrane for Understanding Structure and Proton Conductivity at Various Hydration Levels Using Neural Network Potential
Attila Taborosi, Kentaro Aoki, Nobuyuki Zettsu, Michihisa Koyama, Yuki NagaoAlkyl sulfonated polyimides (ASPIs), as alternative polymer electrolytes for fuel cells, are known to exhibit lyotropic liquid crystalline behavior upon water uptake, forming organized lamellar structures and achieving high proton conductivity. Previous experimental studies have shown that ASPIs with planar backbones exhibit enhanced proton conductivity (0.2 S/cm) compared to those with bent backbones (0.03 S/cm). To explain this difference at the atomistic level, molecular dynamics simulations were conducted using a universal neural network potential. The appearance of monomer unit length in planar ASPIs, indicating higher molecular order, was found to correlate with higher proton conductivity compared to that of bent ASPIs. Despite the similar deprotonation and solvation of sulfonic acid groups in both planar and bent ASPIs, the proton conductivity was independent of these factors. Directional mean square displacement analysis provided further insights into the differences in proton conductivity between planar and bent types.
Read moreComparison of Computational Methods for Simulating Depolymerization Reaction
Shunsuke MiedaA chemical recycling process that reduces polymers to their raw materials plays a crucial role in circular economy. To contribute to chemical recycling, this study proposes a system that simulates the process of depolymerization from polymer-to-monomer using reactive molecular dynamics (MD). Two MD methods, Reax force field (ReaxFF) and neural network potential (NNP), were employed to simulate the depolymerization of a polystyrene model. We validated the simulation accuracies by comparing monomer yields and decomposition products with experimental results. The results showed that NNP-MD accurately replicated the degradation and redecomposition processes and achieved consistency with the experimental data at various temperatures. ReaxFF-MD, however, was less able to represent the depolymerization process. We conclude that NNP-MD is capable of simulating polymer depolymerization results that are consistent with experimental observations. These results contribute to the development of methods for efficient chemical recycling and the broader realization of a circular economy.
Read moreDefect-Driven Evolution of Oxo-Coordinated Cobalt Active Sites with Rapid Structural Transformation for Efficient Water Oxidation
Jinseok Koh, Choah Kwon, Hyunjeong Kim, Eunchong Lee, Akihiko Machida, Yuki Nakahira, Yun Jeong Hwang, Kouji Sakaki, Sangtae Kim, Eun Seon ChoReconstructing the surface nature of metal–organic frameworks (MOFs) as precatalytic structures is a promising methodology for improving electrocatalytic performance. However, regulating the structural evolution of MOFs during electrolysis remains highly uncontrollable and lacks an in-depth understanding of the role of in situ-derived active sites. Here, we suggest a simple approach to fine-tune the symmetry of Co-MOFs with an oxo-coordinated asymmetric coordination that acts as a prototypical structure motif for the oxygen evolution reaction (OER). Through a facile thermal treatment, the Co–N4 configuration of Co-MOFs transforms to the distorted Co–N3–oxo configuration of defective Co–ligand nanoclusters. By operando spectroscopic characterization, the reconstructed Co–N3–oxo structure enables a rapid structural transition toward homogeneous oxyhydroxides. Moreover, the defective nature of the precatalytic structure regulates the surface Co–O bonding environment with abundant μ2-O–Co3+ sites, thereby exhibiting highly enhanced OER activity with an overpotential of 256 mV at 10 mA cm–2 and excellent durability for 100 h, compared with the pristine Co-MOFs. Atomistic simulations reveal that the effect of OER intermediates on the oxyhydroxides gets distributed among neighboring Co ions, promoting balanced binding of the intermediates. This work highlights an effective strategy to design the MOF-based structure for optimizing the surface nature, thus enhancing the electrocatalytic activity.
Read moreTunable Ion Energy Barrier Modulation through Aliovalent Halide Doping for Reliable and Dynamic Memristive Neuromorphic Systems
Jongmin Bae, Choah Kwon, See-On Park, Hakcheon Jeong, Taehoon Park, Taehwan Jang, Yoonho Cho, Sangtae Kim, Shinhyun ChoiMemristive neuromorphic computing has emerged as a promising computing paradigm for the upcoming artificial intelligence era, offering low power consumption and high speed. However, its commercialization remains challenging due to reliability issues from stochastic ion movements. Here, we propose an innovative method to enhance the memristive uniformity and performance through aliovalent halide doping. By introducing fluorine concentration into dynamic TiO2−x memristors, we experimentally demonstrate reduced device variations, improved switching speeds, and enhanced switching windows. Atomistic simulations of amorphous TiO2−x reveal that fluoride ions attract oxygen vacancies, improving the reversible redistribution and uniformity. A number of migration barrier calculations statistically show that fluoride ions also reduce the migration energies of nearby oxygen vacancies, facilitating ionic diffusion and high-speed switching. The detailed Voronoi volume analysis further suggests design principles in terms of the migrating species’ electrostatic repulsion and migration barriers. This work presents an innovative methodology for the fabrication of reliable memristor devices, contributing to the realization of hardware-based neuromorphic systems.
Read moreConstructing Reversible Li Deposition Interfaces by Tailoring Lithiophilic Functionalities of Heteroatom-Doped Graphene Interlayer for Highly Stable Li Metal Anodes
Beom Gwon Son, Choah Kwon, YongJun Cho, Taegyu Jang, Hye Ryung Byon, Sangtae Kim, Eun Seon ChoLithium (Li) metal has been regarded as the ideal anode for rechargeable batteries due to its low reduction potential and high theoretical capacity. However, the formation of fatal Li dendrites during repeated cycling shortens the battery life and causes serious safety concerns. Functionalized separators with electrically conductive and lithiophilic coating layers potentially inhibit dendrite formation and growth on Li metal anodes by providing nucleation sites for reversible Li deposition/stripping. In this work, we propose functionalized separators incorporating heteroatom-doped (N or B) graphene interlayers to modulate the Li nucleation behavior. The electronegative N-doping and electropositive B-doping were investigated to understand their regulation of the Li deposition behavior. With the heteroatom-doped graphene-coated separators, we observe significantly improved cycling stability along with enhanced charge transfer kinetics and low Li nucleation overpotential. This is attributed to the heteroatom-doped graphene interlayer expanding the surface area of the Li metal anode while providing additional space for uniform Li deposition/stripping, thus preventing undesirable side reactions. As a result, the formation of dendrites and pits on the Li metal anode surface is suppressed, demonstrating the protective effect of the Li metal anode. Interestingly, N-doped graphene-coated separators exhibit lower Li nucleation overpotentials than B-doped graphene-coated separators but rather lower average Coulombic efficiencies and reduced cycling stability. This implies that adequate adsorption on B-based sites, as opposed to the strong adsorption on N-based sites, improves the reversibility. Notably, the Li adsorption strength of the lithiophilic functional groups critically affects the reversibility, as observed by Li nucleation barrier measurements and atomistic simulations. This work suggests that interface engineering using conductive and lithiophilic materials can be a promising strategy for controlling Li deposition in advanced Li metal batteries.
Read moreSustained Area-Selectivity in Atomic Layer Deposition of Ir Films: Utilization of Dual Effects of O3 in Deposition and Etching
Han Kim, Taeseok Kim, Hong Keun Chung, Jihoon Jeon, Sung-Chul Kim, Sung Ok Won, Ryosuke Harada, Tomohiro Tsugawa, Sangtae Kim, Seong Keun KimArea-selective deposition (ASD) based on self-aligned technology has emerged as a promising solution for resolving misalignment issues during ultrafine patterning processes. Despite its potential, the problems of area-selectivity losing beyond a certain thickness remain critical in ASD applications. This study reports a novel approach to sustain the area-selectivity of Ir films as the thickness increases. Ir films are deposited on Al2O3 as the growth area and SiO2 as the non-growth area using atomic-layer-deposition with tricarbonyl-(1,2,3-η)-1,2,3-tri(tert-butyl)-cyclopropenyl-iridium and O3. O3 exhibits a dual effect, facilitating both deposition and etching. In the steady-state growth regime, O3 solely contributes to deposition, whereas in the initial growth stages, longer exposure to O3 etches the initially formed isolated Ir nuclei through the formation of volatile IrO3. Importantly, longer O3 exposure is required for the initial etching on the growth area(Al2O3) compared to the non-growth area(SiO2). By controlling the O3 injection time, the area selectivity is sustained even above a thickness of 25 nm by suppressing nucleation on the non-growth area. These findings shed light on the fundamental mechanisms of ASD using O3 and offer a promising avenue for advancing thin-film technologies. Furthermore, this approach holds promise for extending ASD to other metals susceptible to forming volatile species.
Read moreAtom-by-atom design of Cu/ZrOx clusters on MgO for CO2 hydrogenation using liquid-phase atomic layer deposition
Seongmin Jin, Choah Kwon, Aram Bugaev, Bartu Karakurt, Yu-Cheng Lin, Louisa Savereide, Liping Zhong, Victor Boureau, Olga Safonova, Sangtae Kim, Jeremy S. LuterbacherThe difficulty of synthesizing uniform atomically precise active sites limits our ability to engineer increasingly more active heterogeneous catalysts for the hydrogenation of CO2 to methanol. Here we design Cu/ZrOx clusters on MgO with near atomic precision for CO2 hydrogenation using a liquid-phase atomic layer deposition method. The controlled cluster structure modulates the binding strength of CO2 and moderately stabilizes monodentate formate—an essential reaction intermediate for methanol production. We achieved a methanol selectivity of 100 and 76.7% at 200 and 250 °C, respectively and a methanol productivity that was one to two orders of magnitude higher than when the same catalysts were prepared by impregnation. Ab initio computations show that Cu/ZrOx clusters can tune the oxidation of Zr, which controls the stability of reaction intermediates on the catalyst. Our approach demonstrates the potential of precise atomic control of catalytic clusters to improve catalytic productivity.
Read moreFacile Synthesis of Nanoporous Mg Crystalline Structure by Organic Solvent-Based Reduction for Solid-State Hydrogen Storage
Hyesun Kim, HyeonJi Kim, Wonsik Kim, Choah Kwon, Si-Won Jin, Taejun Ha, Jae-Hyeok Shim, Soohyung Park, Aqil Jamal, Sangtae Kim, Eun Seon ChoNanoporous metals have unique potentials for energy applications with a high surface area despite the percolating structure. Yet, a highly corrosive environment is required for the synthesis of porous metals with conventional dealloying methods, limiting the large-scale fabrication of porous structures for reactive metals. In this study, we synthesize a highly reactive Mg nanoporous system through a facile organic solution-based approach without any harsh etching. The synthesized nanoporous Mg also demonstrates enhanced hydrogen sorption kinetics and reveals unique kinetic features compared to Mg nanoparticles. The well-crystallized Mg nanoporous structure exhibits crystalline facet-dependent hydrogen sorption characteristics, featuring gradually improved hydrogen storage capacity up to 6 wt.% upon cycling. Also, continuum kinetics models coupled to atomistic simulations reveal that the compressive stress developed during the hydrogenation of nanoporous Mg enhances the sorption kinetics, as opposed to the sluggish kinetics under tensile stress in core-shell nanoparticles. It is expected that the synthetic strategy conceived in this study can be further implemented to prepare different kinds of reactive porous metals in a facile and scalable way for the development of large-scale and distributed hydrogen storage systems for the emerging low-carbon hydrogen economy.
Read moreInterconnected Lamellar 3D Semiconductive PCP for Rechargeable Aqueous Zinc Battery Cathodes
Zirui Lin, Ken-ichi Otake, Takashi Kajiwara, Shotaro Hiraide, Maryam Nurhuda, Daniel Packwood, Kentaro Kadota, Hirotoshi Sakamoto, Shogo Kawaguchi, Yoshiki Kubota, Ming-Shui Yao, Satoshi Horike, Xiaoqi Sun, Susumu Kitagawa2D electronically conductive porous coordination polymers/metal–organic frameworks (2D EC-MOFs) of M-HHTPs (HHTP = 2,3,6,7,10,11-hexahydroxytriphenylene; M = Co, Ni, Cu, etc.) have received extensive attention due to their ease of preparation, semiconductive properties, and tunability based on the choice of metal species. However, slight shifts between layers attenuate their specific surface area and stability. In this study, the metal-ion bridge strategy is newly adopted and a vanadyl counterpart of M-HHTP is synthesized with a chemical formula of (VO)3(HHTP)2, hereafter referred to as VO-HHTP. The semiconductor VO-HHTP has a vertical interconnection by octahedral VO6 chains and exhibits a relatively high specific surface area (ca. 590 m2 g−1) compared to other 2D EC-MOFs. Motivated by its redox activity and porous nature, VO-HHTP is applied as the cathode material in rechargeable aqueous zinc batteries (RAZBs). VO-HHTP demonstrates a high capacity of 240 mAh g−1 and excellent rate capability, even with a reduced amount of conductive agent, surpassing the performance of the previous EC-MOFs. Furthermore, its stable structure ensures long-term cycling stability, addressing a common issue in previous EC-MOFs. The work contributes to the development of new concepts in both the design of π-conjugated EC-MOFs and the study of cathode materials for RAZBs.
Read moreDirect derivation of anisotropic atomic displacement parameters from molecular dynamics simulations demonstrated in thermoelectric materials
Yoyo HinumaAtomic displacement parameters (ADPs) are crystallographic information that describe the statistical distribution of atoms around an atom site. Direct derivation of anisotropic ADPs by atom from molecular dynamics (MD) simulations, where the (co)valences of atom positions are taken over recordings at different time steps in a single MD simulation, was demonstrated on three thermoelectric materials, Ag8SnSe6, Na2In2Sn4, and BaCu1.14In0.86P2. Unlike the very frequently used lattice dynamics approach, the MD approach can obtain ADPs in disordered crystals and at finite temperature, but not under conditions where atoms migrate in the crystal. ADPs from MD simulations would act as a tool complementing experimental efforts to understand the crystal structure including the distribution of atoms around atom sites.
Read moreStructural Stabilization and Superior Electrochemical Performance of Yttrium-Doped O₃-NaFe₀.₄Ni₀.₃Mn₀.₃O₂ Cathodes for Sodium-Ion Batteries
Tingru Chen, Eugenio H. Otal, Tien Quang Nguyen, Shunsuke Narumi, Michihisa Koyama, and Nobuyuki ZettsuThis is the first demonstration of successful Y3+ doping, and Y3+-doped O3-NaFe0.4Ni0.3Mn0.3O2 (NaFNM) cathodes exhibit superior cyclability and C-rate capabilities. Y3+ doping enabled cells with 91% capacity retention at 1 C after 100 cycles and a high specific capacity of 102 mA h g–1 at a 7 C rate. Furthermore, the full cell delivered outstanding cycling stability, with a capacity retention of 80% after 500 cycles at 1 C and 71.3% after 1000 cycles. Operando X-ray diffraction spectroscopy (XRD) and X-ray absorption spectroscopy, along with simulations using density functional theory and neural network potential methods, provided comprehensive insights on the effects of Y3+ doping on the cycling characteristics. Our results confirm that Y3+ doping effectively stabilizes the O3 structure, enhances the electrochemical performance, and addresses the challenge of irreversible O3 to P3 phase transitions.
Read moreNeural Network Potential Molecular Dynamics Simulations of (La,Ce,Pr,Nd)0.95(Mg,Zn,Pb,Cd,Ca,Sr,Ba)0.05F2.95
Yoyo HinumaTysonite structure fluorides doped with divalent cations, represented by Ce0.95Ca0.05F2.95, are a class of good F– ion conductors together with fluorite-structured compounds. Computational understanding of the F– conduction process is difficult because of the complicated interactions between three symmetrically distinct F sites and the experimentally observed change in the F diffusion mechanism slightly above room temperature, effectively making first-principles molecular dynamics (FP-MD) simulations, which are often conducted well above the transition temperature, useless when analyzing behavior below the transition point. Neural network potential (NNP) MD simulations showed that the F diffusion coefficient is higher when the divalent dopant cation size is similar to the trivalent cation size. The diffusion behavior of F in different sites changes at roughly 500 K in Ce0.95Ca0.05F2.95 because only the F1 site sublattice contributes to F diffusion below this temperature, but the remaining F2 and F3 sublattices become gradually active above this temperature. The paradox of higher diffusion coefficients in CeF3-based compounds than similar LaF3-based compounds even though the lattice parameters are larger in the latter may be caused by a shallower potential of Ce and F in CeF3 compared to the LaF3 counterparts.
Read moreMolecular dynamics study of the effect of composition on elastic properties of silicon oxynitride films
Sakurako Miyazaki, Hiroki Sakakima, Keigo Ogawa, Satoshi IzumiUnderstanding the mechanical properties of silicon oxynitride (a-SiON), a key insulating material, is vital for electronic device design and reliability. Though the effects of fabrication conditions on a-SiON have been studied, the underlying relationship between its atomic-scale structure and mechanical properties remains unclear. This study investigates the relationship between elasticity and atomic-scale structures in a-SiON by molecular dynamics simulations with a universal graph neural network interatomic potential. The bulk modulus increases from 49 to 150 GPa with higher N content. N atoms form N2 molecules under O-rich conditions, hindering bulk modulus increase, and form an Si3N4-like network under O-poor conditions, enhancing bulk modulus. Formation energy calculations indicate N2 formation is preferable under O-rich conditions. Meanwhile, under O-poor conditions, Si–N bond formation is preferable, which reinforces a-SiON by increasing bond density. The findings suggest realizing O-poor conditions is crucial for highly elastic insulating films.
Read moreMolecular Dynamics of Catalyst-Free Edge Elongation of Boron Nitride Nanotubes Coaxially Grown on Single-Walled Carbon Nanotubes
Kaoru Hisama, Ksenia V. Bets, Nitant Gupta, Ryo Yoshikawa, Yongjia Zheng, Shuhui Wang, Ming Liu, Rong Xiang, Keigo Otsuka, Shohei Chiashi, Boris I. Yakobson, Shigeo MaruyamaRecent advances in low-dimensional materials have enabled the synthesis of single-walled carbon nanotubes encapsulated in hexagonal boron nitride (BN) nanotubes (SWCNT@BNNT), creating one-dimensional van der Waals (vdW) heterostructures. However, controlling the quality and crystallinity of BNNT on the surface of SWCNTs using chemical vapor deposition (CVD) remains a challenge. To better understand the growth mechanism of the BNNT in SWCNT@BNNT, we conducted molecular dynamics (MD) simulations using empirical potentials. The simulation results suggest that spontaneous BN nucleation is unlikely to occur on the outer surface of the SWCNT when we assume only vdW interaction between the BN and SWCNT layers. However, we observe the elongation of the BNNT when a short BNNT is provided as a seed nucleus on the SWCNT. This grown BNNT structure, with its sharply cut edges, aligns with experimental observations made using transmission electron microscopy (TEM). Moreover, the edge-reconstruction process favors zigzag B edges, which exhibit low edge energy according to the ReaxFF potential. Our simulation successfully provides insights into the catalyst-free growth process of this one-dimensional vdW heterostructure.
Read moreSolid-liquid phase boundary of oxide solid solutions using neural network potentials
Kazushige Hyodo, Kenta Hongo, Tom Ichibha, Ryo MaezonoWe propose a cost-effective computational approach for predicting the phase boundary of oxide solid solutions, i.e., melting point, by identifying the point where the free energies of the solid and liquid phases are equivalent. To evaluate the melting point, we computed the temperature-dependent free energies of both phases using the thermodynamic integration (TI) method. This was combined with the reverse-scaling technique, employing the Einstein model for the solid and the scaled Uhlenbeck–Ford model for the liquid as the reference systems. The change in free energy between the real and reference states in the present TI method was calculated as the ensemble average of the configurations sampled from molecular dynamics (MD) simulations using neural network potentials (NNPs) trained on first-principles density functional theory (DFT) data. Initially, we benchmarked copper (Cu) as a typical metal. Tungsten (W) and magnesium oxide (MgO) were then tested as typical high melting-point metals and oxides, respectively, achieving good agreement with both ab initio MD (AIMD) results and experimental data. We then applied our established approach to a BaO–CaO oxide solid solution, observing that the computed phase boundary aligns well with Calphad predictions from previous studies. The integration of NNPs into phase boundary prediction is essential for reducing computational costs while ensuring accuracy, compared with AIMD-based approaches.
Read moreTheoretical Catalyst Screening of Multielement Alloy Catalysts for Ammonia Synthesis Using Machine Learning Potential and Generative Artificial Intelligence
Kaoru Hisama, Atsushi Ishikawa, Susan Menez Aspera, and Michihisa KoyamaIn catalyst design, it is important to use multielement alloys to search for highly reactive atomic compositions and configurations. We developed a rapid calculation method for finding highly reactive surfaces by generating candidates of atomic configurations with high activity using a generative adversarial network (GAN). The evaluation of the reaction energy and turnover frequency (TOF) for each catalyst candidate is necessary; however, this is computationally costly with conventional methods such as density functional theory (DFT). Here, this was accelerated by the universal neural network potential (UNNP). By iterating the generation and evaluation processes, we explored atomic configurations that improve the reactivity for the ammonia synthesis of an alloy catalyst comprising Pd, Rh, and Ru. The microkinetic analysis, considering the six elementary reactions in the NH3 synthesis, was performed on more than 600 catalyst candidates. The generated ternary alloy surface structures exhibited higher TOFs than their binary and monometallic counterparts. The highly reactive configurations exhibited stronger N adsorption on the Ru–Ru bridge site on the step of the surface surrounded by Pd atoms. This method can be employed for an efficient computational catalyst screening, with detailed atomistic structure of catalysts and thermodynamics or kinetics properties.
Read moreEffects of Alkyl Side Chain Length on the Structural Organization and Proton Conductivity of Sulfonated Polyimide Thin Films
Tetsuya Honbo, Yutaro Ono, Kota Suetsugu, Mitsuo Hara, Attila Taborosi, Kentaro Aoki, Shusaku Nagano, Michihisa Koyama, and Yuki NagaoThis study investigates the impact of alkyl side chain length on the structural organization and proton conductivity of sulfonated polyimide (SPI) thin films. SPIs with different alkyl sulfonated side chain lengths CX (X: number of carbon atoms at the side chain, X = 0, 3, 6, and 10) were synthesized, and the relationship between molecular architecture and proton transport properties was investigated. In all SPIs, the polymer backbone was oriented parallel to the substrate, and the lamellar structures were confirmed, with spacing increasing linearly with side chain length. Water uptake behavior and proton conductivity varied significantly, where the C0 thin film exhibited the highest water uptake and proton conductivity at lower humidity while the C3 thin film achieved higher conductivity under high humidity, reaching 1.8 × 10–1 S cm–1 at 298 K and 95% relative humidity (RH) and an activation energy of 0.18 eV at 90% RH. Conversely, extending the alkyl side chain length led to the insolubility in water in C10. As a result, a proton exchange membrane system with high chemical/water stability and high proton conductivity was constructed. These findings provide critical insights into designing advanced proton-conducting materials and optimizing their performance for fuel cells and other electrochemical devices.
Read moreRise of machine learning potentials in heterogeneous catalysis: Developments, applications, and prospects
Seokhyun Choung, Wongyu Park, Jinuk Moon, Jeong Woo HanThe urgency of tackling climate change is driving a global shift towards renewable sources of energy, with a growing contribution from alternative energy sources such as solar, wind and hydroelectric power. With the global push for the sustainable energy, the demand for effective catalysts for sustainable chemical production and energy storage has been rapidly increasing. Computational simulations have contributed to the rational design of catalysts by allowing profound analysis of catalyst properties. Machine learning potential (MLP) has emerged as a potential tool to bridge the gap between quantum mechanical accuracy and computational efficiency, overcoming the computational cost limitations of quantum chemistry-based simulations. This review discusses the development and application of MLP in multiscale simulations of heterogeneous catalysis. It covers the basic concepts of computational catalysis, the construction of MLP focusing on efficient datasets, atomic structure representations, and the process of training and evaluating of ML models. Furthermore, the potential applications of MLP are discussed in addressing computational challenges within the field, as MLP has potential to overcome limitations in simulation time and length scale. Lastly, the prospects for MLP are presented, taking advantage of the rapid advancements in artificial intelligence architectures. It is expected that the integration of MLP will accelerate progress within the catalyst research community and will bridge the gap between theoretical and experimental approaches in catalytic research.
Read moreIntelligent Stress-Adaptive Binder Enabled by Shear-Thickening Property for Silicon Electrodes of Lithium-Ion Batteries
Ohhyun Kwon, Tae Yong Kim, Taewon Kim, Jihyeon Kang, Seohyeon Jang, Hojong Eom, Seyoung Choi, Junhyeop Shin, Jongkwon Park, Myeong-Lok Seol, Jeong Woo Han, Soomin ParkElastic binders with supramolecular interactions are widely explored to mitigate the stress caused by the volume expansion of electrode materials, such as Si, S, or Li metals, in next-generation secondary batteries. Herein, a new class of elastic binders is proposed with an automatic stress-control mechanism capable of responding in real time to dynamic local stress variations. Specifically, this study focuses on the shear-thickening behavior, wherein polymers automatically amplify their viscoelasticity in response to local shear-stress changes. To realize an intelligent stress-adaptive binder, starch analogs exhibiting shear-thickening properties and unique crystallinity are employed as binders for highly expandable Si anodes. The shear-thickening mechanism is comprehensively investigated using deep-learning-based molecular dynamics (MD) simulations and in situ transmission electron microscopy (TEM) analysis, which determines the optimal conditions for effectively limiting dynamic local surface expansion. Among the starch analogs, the amylose and long-chain amylopectin (AMLAP) binder demonstrates improved high-rate capability (1710 mAh g−1 at 5 C) and superior reversible capacity (2025 and 1493 mAh g−1 after 100 and 500 cycles, respectively, at 1 C) with optimal shear-thickening properties. Furthermore, AMLAP exhibits favorable characteristics for affordable large-scale production. Hence, this study clearly demonstrates that the shear-thickening properties of binders can be considered a new factor in fabricating stable electrodes with extremely expandable materials.
Read moreExperimental study on Na⁺ conductivity in NaAlBr4 and atomic-scale investigation of Na+ conduction
Reona Miyazaki, Masanobu Nakayama, Takehiko HiharaThe ionic conduction properties of Li/Na metal halides have been extensively studied, with recent attention turning towardsAl-based systems. However, limited studies have focused on alkali Al bromides. In this study, we explored the Na+ conductionproperties of NaAlBr4 . Conductivity measurements at 30 °C revealed a Na + conductivity of 1.2 × 10 −5 S/cm, surpassing thatof isostructural NaAlCl4 threefold. Molecular dynamics (MD) simulations to elucidate the conduction mechanisms revealedthat Na + conduction was not observed in stoichiometric NaAlBr4 , which has high formation energies of Na + vacancies andinterstitials (0.88 eV and 0.73 eV, respectively). Nevertheless, a conductivity of 1.2 × 10−5 S/cm was observed. The activationenergy for ion conduction was experimentally determined as 0.43 eV, and the migration energies were calculated as 0.26 eV(Na + vacancies) and 0.16 eV (Na + interstitials) by MD simulations. These discrepancies in ion conduction were partiallyexplained by the role of transient defects enriched via ball milling in facilitating Na + conduction on the particle surface,offering insights into the complex ion conduction of ball-milled NaAlBr4 .
Read moreExploring Electrolyte Adsorption on the Different Types of Layered Cathode Surfaces in Lithium-Ion Batteries via a Universal Neural Network Potential Method
Attila Taborosi, Hiromasa Shiiba, Michihisa Koyama, and Nobuyuki ZettsuLiNixMnyCozO2 (NCM) is a promising cathode material for lithium-ion batteries. Their long-term stability and impedance growth as cycles are strongly influenced by the properties of the cathode electrolyte interface (CEI), formed by the reaction between the electrolyte and electrode surface. Understanding of these interactions at the atomic level of the NCM electrode surface and electrolyte will provide a new strategic approach for the design of a highly functional CEI layer. In this study, we explored the influence of Ni content in transition metal layers on surface energies under different synthetic conditions and terminations using a density functional theory (DFT) validated universal neural network potential (UNNP) method. Furthermore, we investigated the adsorption of ethylene carbonate (EC) and dimethyl carbonate (DMC) on the most favorable NCM surfaces. EC and DMC displayed similar adsorption energy trends; however, differences were observed in the preferred configurations, which can affect the formation of the CEI.
Read moreUniversal Neural Network Potential-Driven Molecular Dynamics Study of CO2/O2 Evolution at the Ethylene Carbonate/Charged–Electrode Interface
Motoki Horibe, Naoto Tanibata, Hayami Takeda, and Masanobu NakayamaLong-term durability and safety are required to develop Li-ion batteries that can operate at high voltages. However, side reactions, including the release of O2 from the electrode and CO2 from the organic electrolyte, occur at the positive-electrode/electrolyte interface during charging at high voltages. In this study, universal neural network potential (UNNP)-driven molecular dynamics (MD) calculations are used to investigate the mechanism of the reaction between LixCoO2 (0 ≤ x ≤ 1) or LixNiO2 (0 ≤ x ≤ 1), as the positive-electrode material, and an ethylene-carbonate-based electrolyte, with a solid–liquid interface composed of ∼1700 atoms. Molecular CO2 and O2 evolve from the partially or fully Li-deintercalated LixNiO2, while no gas–evolution reactions are observed for LixCoO2. Hence, compared LixNiO2, the LiCoO2 electrode is more stable toward the decomposition of ethylene carbonate in the charged state. The decomposition reactions at the solid–liquid interface during charging are also analyzed using a NN force field. This study provides a robust approach involving MD simulations using UNNP to better understand the side reactions in electrochemical devices, which can guide manufacturers in selecting appropriate materials.
Read moreStress-Induced Martensitic Transformation in Na3YCl6
Akira Miura, Koki Muraoka, Kotaro Maki, Saori Kawaguchi, Kazuhiro Hikima, Hiroyuki Muto, Atsunori Matsuda, Ichiro Yamane, Toshihiro Shimada, Hiroaki Ito, Yoshikazu Mizuguchi, Chikako Moriyoshi, Hiroshi Nakajima, Shigeo Mori, Hiroshi Oike, Akira Nakayama, Wenhao Sun, Nataly Carolina Rosero-Navarro, Kiyoharu TadanagaMartensitic transformation with volume expansion plays a crucial role in enhancing the mechanical properties of steel and partially stabilized zirconia. We believe that a similar concept could be applied to unexplored nonoxide materials. Herein, we report the stress-induced martensitic transformation of monoclinic Na3YCl6 with an ∼3.4% expansion. In situ synchrotron X-ray diffraction and atomistic simulations showed that anisotropic crystallographic transformation from monoclinic to rhombohedral Na3YCl6 occurs exclusively under uniaxial pressure; no effect is observed under hydrostatic pressure conditions. The uniaxially pressed powder compact of monoclinic Na3YCl6 showed a large indentation impression and low Young’s modulus, in contrast to its high bulk modulus, suggesting that these unique mechanical properties are induced by the martensitic transformation.
Read moreEffect of very slow O diffusion at high temperature on very fast H diffusion in the hydride ion conductor LaH2.75O0.125
Yoyo HinumaNeural network potential based molecular dynamics (MD) simulations on the excellent H conductor LaH2.75O0.125 show that O starts diffusing above a critical temperature of Tc ∼ 550 K, according to the variance of atom positions regardless of the time step. The original diffusion process at temperatures below Tc has an activation barrier of 0.25 eV. Use of MD simulations with various O and La mass revealed, at above Tc, the coexistence of the 0.25 eV process and an additional diffusion process with an activation barrier of 0.20 eV. The O and La have strongly anharmonic characters.
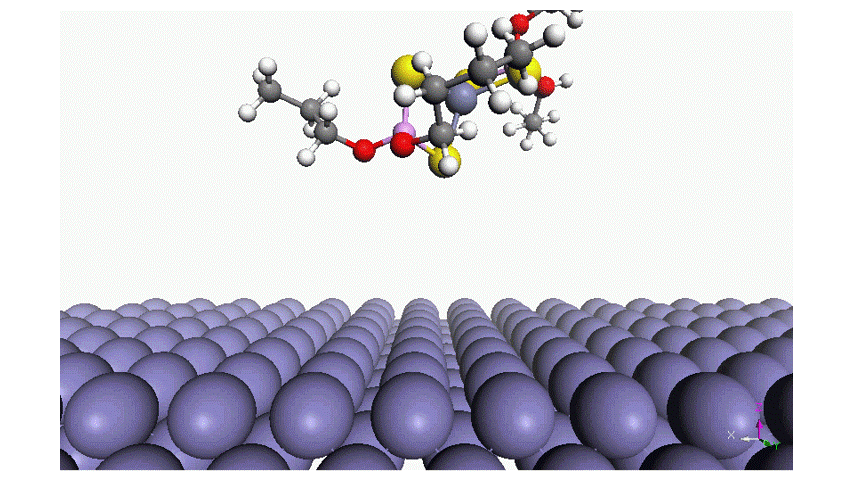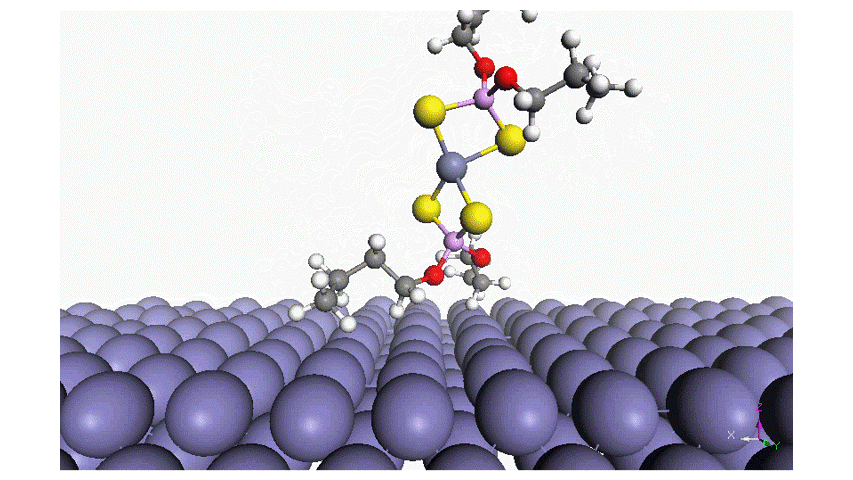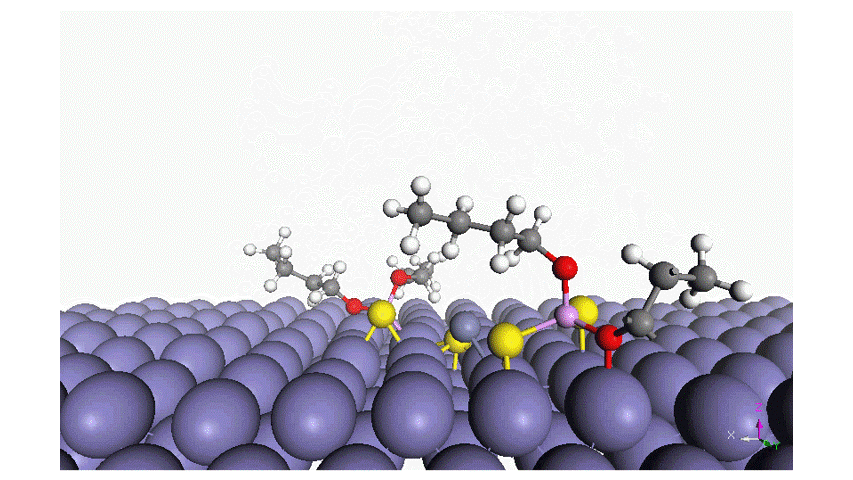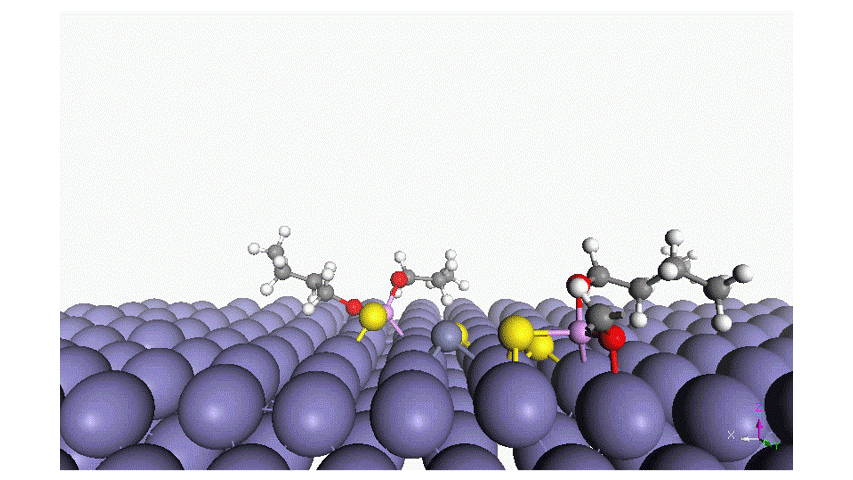
Read moreAdsorption of Phosphorus-Type Anti-Wear Agents for Refrigerator Nonpolar-Oil Additives
Hiromi YUASA , Tomohiro TAKAKI , Tasuku ONODERA , Yuji SHITARA , Kyosuke ARAKAWA , Masaaki AKAMATSU, Hideki SAKAI and Kenichi SAKAIIn this study, we characterized the adsorption of several phosphorus-type anti-wear agents on an iron oxide surface in hexane. The phosphorus-type anti-wear agents employed in this study were triphenyl phosphate and its derivatives. The quartz crystal microbalance with dissipation monitoring technique revealed that the adsorption of the phosphorus-type anti-wear agents was strongly dependent on their chemical reactivity; i.e., the anti-wear agents with reactive functional groups result in large adsorption amount on the iron oxide surface. Additionally, (i) the polarity of the anti-wear agents as well as (ii) the presence of a benzene ring in their chemical structure may also affect the adsorption behavior. Adsorption energy of the anti-wear agent was also estimated by an atomistic level simulator “MATLANTISTM” and the simulation results well supported the experimental findings. Four-ball tests further demonstrated that the chemical or physical adsorption film formed on the iron oxide surface resulted in the wear resistance, as expected. This study provides fundamental knowledge on the structure-performance relationship of phosphorus-type anti-wear agents, giving future perspectives for lubricant formulations.
Read moreIn situ ambient pressure x-ray photoelectron spectroscopy study on O2/H2O-assisted Na–CO2 batteries
Kevin Iputera , Chia-Hui Yi , Jheng-Yi Huang, Masanobu Nakayama, Bo-Hong Liu, Chia-Hsin Wang, Yaw-Wen Yang, Ru-Shi LiuNa–CO2 battery is considered a high-energy-density storage system with the ability to utilize CO2. However, unlike its Li counterpart, it suffers from an unclear reaction mechanism and poor battery performance. CO2 is an inert species and requires high activation energy. Moreover, the understanding of how additives such as O2 and H2O aid the cathode reaction remains lacking. Herein, the mechanism of the CO2 reduction reaction is unraveled through in situ ambient pressure X-ray photoelectron spectroscopy (APXPS). The oxidation states of the discharged products can be well revealed to understand the reactions. Unlike previous studies, a pure CO2 environment was found to have poor electrochemical activity. When additives, such as O2 and H2O, were introduced to the system, some Csp2 was formed. We propose that Csp2 should be attributed to the alkene formation as the decomposition product of ionic liquid, which serves as the electrolyte. The formation of elemental carbon as the discharge product is unlikely, which is in stark contrast to previous studies. As a result, the poor electrochemical activity of the pure CO2 system leads to the formation of CO, which escapes from the electrode surface and results in poor reversibility. Additives such as O2 and H2O are electrochemically active themselves, while CO2 participated in the reaction chemically, reducing the chemical reversibility. Therefore, a system without CO2 is more beneficial to the Na–air batteries.
Read moreHydrogen production by NH3 decomposition at low temperatures assisted by surface protonics
Yukino Ofuchi, Kenta Mitarai, Sae Doi, Koki Saegusa, Mio Hayashi, Hiroshi Sampei, Takuma Higo, Jeong Gil Seo and Yasushi SekineAmmonia, which can be decomposed on-site to produce CO2-free H2, is regarded as a promising hydrogen carrier because of its high hydrogen density, wide availability, and ease of transport. Unfortunately, ammonia decomposition requires high temperatures (>773 K) to achieve complete conversion, thereby hindering its practical applicability. Here, we demonstrate that high conversion can be achieved at markedly lower temperatures using an applied electric field along with a highly active and readily producible Ru/CeO2 catalyst. Applying an electric field lowers the apparent activation energies, promotes low-temperature conversion, and even surpasses equilibrium conversion at 398 K, thereby providing a feasible route to economically attractive hydrogen production. Experimentally obtained results and neural network potential studies revealed that this reaction proceeds via HN–NH intermediate formation by virtue of surface protonics.
Read moreMechanistic Study on NH3 Decomposition over Cu-Exchanged CHA Zeolites Using Automated Reaction Route Mapping Combined with Neural Network Potential and Density Functional Theory Calculations
Shunsaku Yasumura, Taisetsu Kato, Yucheng Qian, Takashi Toyao, and Ken-ichi ShimizuThe reaction mechanism for the decomposition of NH3 into N2 by Cu-exchanged CHA zeolite (Cu-CHA) was explored by using a combination of neural network potential (NNP) and automated reaction route mapping. According to the revealed reaction route, the formation of N2H4, which thermally decomposes into N2 and H2, is not assisted by both isolated Cu2+ cation and [Cu(OH)]+ species because of their high energy barriers (>300 kJ/mol). In contrast, [Cu(OH)]+ species easily generates [Cu(NH2)]+ species at a much lower energy barrier of 70 kJ/mol. Then, reaction route mapping was performed using two [Cu(NH2)]+ species at Al sites close to each other, resulting in a lower activation barrier than that obtained using only one [Cu(NH2)]+ species to generate N2H4 intermediates (Ea = 119 kJ/mol). In situ X-ray absorption spectroscopy (XAS) experimentally verified that the proximity of the Cu species in zeolite is crucial in suppressing the decomposition of NH3.
Read moreA Practical and Sustainable Ni/Co-Free High-Energy Electrode Material: Nanostructured LiMnO2
Yuka Miyaoka, Takahito Sato, Yuna Oguro, Sayaka Kondo, Koki Nakano, Masanobu Nakayama, Yosuke Ugata, Damian Goonetilleke, Neeraj Sharma, Alexey M. Glushenkov, Satoshi Hiroi, Koji Ohara, Koji Takada, Yasuhiro Fujii, Naoaki YabuuchiNi/Co-free high-energy positive electrode materials are of great importance to ensure the sustainability of Li-ion battery production and its supply chain in addition to minimizing environmental impact. Here, nanostructured LiMnO2 with both orthorhombic/monoclinic layered domains is synthesized, and its lithium storage properties and mechanism are examined. High-energy mechanical milling is used to convert the metastable and nanosized LiMnO2 adopting the cation-disordered rocksalt structure to an optimal domain-segregated layered LiMnO2. This positive electrode produces an energy density of 820 W h kg–1, achieved by harnessing a large reversible capacity with relatively small voltage hysteresis on electrochemical cycles. Moreover, voltage decay for cycling, as observed for Li-excess Mn-based electrode materials, is effectively mitigated. Furthermore, by determining the structure–property relationships of different LiMnO2 polymorphs, LiMnO2 with similar domain structure and surface area is successfully synthesized with an alternative and simpler method, without the metastable precursor and high-energy mechanical milling. The cyclability of domain-containing LiMnO2 is also improved with the use of a highly concentrated electrolyte coupled with a lithium phosphate coating due to the suppression of Mn dissolution. These findings maximize the possibility of the development of high-energy, low-cost, and practical rechargeable batteries made from sustainable and abundant Mn sources without Ni/Co.
Read moreMolecular dynamics of liquid–electrode interface by integrating Coulomb interaction into universal neural network potential
Kaoru Hisama, Gerardo Valadez Huerta, Michihisa KoyamaComputational understanding of the liquid–electrode interface faces challenges in efficiently incorporating reactive force fields and electrostatic potentials within reasonable computational costs. Although universal neural network potentials (UNNPs), representing pretrained machine learning interatomic potentials, are emerging, current UNNP models lack explicit treatment of Coulomb potentials, and methods for integrating additional charges on the electrode remain to be established. We propose a method to analyze liquid–electrode interfaces by integrating a UNNP, known as the preferred potential, with Coulomb potentials using the ONIOM method. This approach extends the applicability of UNNPs to electrode–liquid interface systems. Through molecular dynamics simulations of graphene–water and graphene oxide (GO)–water interfaces, we demonstrate the effectiveness of our method. Our findings emphasize the necessity of incorporating long-range Coulomb potentials into the water potential to accurately describe water polarization at the interface. Furthermore, we observe that functional groups on the GO electrode influence both polarization and capacitance.
Read moreMachine Learning in Catalysis: Analysis and Prediction of CO Adsorption on Multi-elemental Nanoparticle using Metal Coordination-based Regression Model
Susan Menez ASPERA, Gerardo Valadez HUERTA, Yusuke NANBA, Kaoru HISAMA, Michihisa KOYAMAInformation about molecular adsorption strength is important in every catalytic reaction. The ability to compare and determine relevant molecular active sites of interaction is necessary for fast screening of potential catalysts specially in a vast spectrum of probable candidates. In this study, we used the metal-coordination of the adsorption sites as a descriptor of the adsorption energy of CO on the PtRuIr ternary alloy nanoparticle. Using multiple regression model, we are able to predict the adsorption energy and specify some important descriptors that controls the strength of CO adsorption energy. This will enable a fast prediction of CO adsorption energy on PtRuIr nanoparticles with varying compositions and possible different morphologies using only the information of the structure of the catalyst. And open up the possibility of predicting adsorption interaction of other combinations of alloys with higher number of metallic compositions for fast screening of appropriate molecule-surface interaction.
Read moreUniversal-neural-network-potential molecular dynamics for lithium metal and garnet-type solid electrolyte interface
Rinon Iwasaki, Naoto Tanibata, Hayami Takeda & Masanobu NakayamaAll-solid-state Li-metal batteries can conceivably improve the safety and extend the driving ranges of electric vehicles. In this regard, the garnet-type solid electrolyte Li7La3Zr2O12 (LLZ) has garnered considerable attention because of its high Li-ion conductivity and nonreactivity towards molten Li metal. Here, we perform molecular dynamics (MD) simulations using a universal neural network potential (UNNP) to analyse the Li-ion exchange at the LLZ/Li interface at the atomic scale. The UNNP-MD calculations show that Li ions traverse the LLZ/Li interface and that excess Li ions relative to the stoichiometric composition accumulate in an approximately 1 nm-thick zone near the LLZ phase interface, signifying the formation of a space-charge layer. Electronic structural analysis of the UNNP-MD-derived configuration, performed using density functional theory calculations, reveals band bending near the LLZ phase interface and the simultaneous suppression of Li metal reduction. These findings can help expedite the development of rationally designed all-solid-state Li-metal batteries.
Read moreOnset of tetrahedral interstitial formation in GaAsN alloys
Cooper, J. J. P., Timothy Jen, A. Novak, Z. Xi, L. Qi, F. U. Naab, Y. Q. Wang, and R. S. GoldmanN incorporation mechanisms in GaAs1−xNx alloys are probed using combined experimental and computational Rutherford backscattering spectrometry and nuclear reaction analysis angular yield scans. For xN < 0.025, in addition to substitutional nitrogen, NAs, (N-N)As, and (N-As)As split-interstitials are observed. However, for xN ≥ 0.025, evidence for N tetrahedral interstitials, Ntetra, emerges. We propose a mechanism for stabilization of Ntetra in which the elastic interaction between Ntetra and NAs is induced by the opposite signs of their misfit volumes. This work opens opportunities for exploring the formation of Ntetra and its influence on the properties of a variety of highly mismatched alloys.
Read moreExploring Ti active sites in Ziegler-Natta catalysts through realistic-scale computer simulations with universal neural network potential
Masaki Fushimi and Devaiah DammaWe investigate the interaction dynamics between TiCl4 molecules and a MgCl2 (110) cluster to understand the complexities of Ziegler-Natta catalysts through molecular dynamics simulations. Our study reveals significant distortion of Mg atoms at the edge of cluster, influencing TiCl4 adsorption behavior, cluster stability, and reactivity. Various TiCl4 configurations on the MgCl2 surface, such as octahedral and bridging forms, illustrate the versatility of surface in accommodating different molecular arrangements, affecting catalytic performance. We explore diverse adsorption sites influenced by surface topology and electronic properties, revealing a range of adsorption energies from 15.7 to 29.2 kcal/mol. This indicates a complex interplay of molecular forces, significantly impacting catalytic efficiency at distorted sites. Nudged Elastic Band calculations reveal varied activation energies for ethylene insertion, suggesting faster reaction rates at certain sites and highlighting highly favorable dynamics at Corradini type Ti active sites. All these findings offer new directions for enhancing catalyst performances.
Read moreQuantitative analysis of plasma-enhanced chemical vapor deposition mechanisms: Quantum chemical and plasma-fluid dynamics investigation on tetraethoxysilane/O2 plasma
Hu Li, Kazuki DenpohThis study aimed to investigate the influence of reactive oxygen species (i.e., neutral O atom and O2+ ion) on deposition rates and film thickness uniformity in tetraethoxysilane (TEOS) plasma, utilizing a combination of plasma-fluid dynamic and quantum chemical (QC) simulations. The plasma simulations employed an improved model based on a previous study [H. Li et al., Jpn. J. Appl. Phys. 58, SEED06 (2019)], specifically tailored for a TEOS/O2/Ar/He gas mixture. In the QC simulations, both flat and step silicon oxide (SiO2) surfaces were employed to investigate the adsorption behavior of SiO molecules, the predominant silicon-containing species in TEOS plasma. These simulations also enabled the examination of the rates of SiO molecule adsorption on SiO2 surfaces, facilitating a direct comparison with the sticking coefficients utilized in the plasma simulation. The results of QC simulations revealed that SiO molecules exhibited a higher energetic preference for adsorption on step surfaces than on flat surfaces, resulting in the formation of new SiOH surface sites. Meanwhile, the plasma simulations demonstrated a strong correlation between the deposition rate and film thickness uniformity and the generation of oxygen species, specifically O atoms and O2+ ions, as well as their respective fluxes. This relationship takes precedence over the influence of TEOS or its fragments colliding with the surface. Notably, higher plasma source frequencies were found to enhance the production of atomic O, which contributed significantly to achieving higher deposition rates.
Read moreAdsorption of dimethylaluminum isopropoxide (DMAI) on the Al2O3 surface: A machine-learning potential study
Miso Kim, Sehee Kim, Bonggeun ShongDimethylaluminum isopropoxide (DMAI) is attracting attention as an alternative precursor for atomic layer deposition (ALD) of aluminum oxide (Al2O3). However, the surface chemical reaction mechanisms of DMAI during ALD regarding its dimeric structure under vacuum deposition process conditions has yet to be clear. In this work, the adsorption mechanism of dimeric and monomeric DMAI on a fully hydroxylated Al2O3 surface is studied using machine-learning potential (MLP) calculations. The initial adsorption of DMAI appears facile and would result in the coexistence of both methyl and isopropoxy ligands on the surface. The reactivity of DMAI is smaller than that of TMA, owing to the propensity of DMAI to adopt a dimeric form. Especially when the substrate is partially covered by other adsorbate species, the large molecular size and low reactivity of dimeric DMAI considerably hinder its reactivity toward surface adsorption. Current results are in good correspondence with the previous experimental results, where lower growth per cycle (GPC) and higher selectivity in area-selective ALD (AS-ALD) could be observed by using DMAI than compared to those of TMA processes.
Read moreHigh-throughput investigation of stability and Li diffusion of doped solid electrolytes via neural network potential without configurational knowledge
Ryohto Sawada, Kosuke Nakago, Chikashi Shinagawa & So TakamotoSolid electrolytes hold substantial promise as vital components of all-solid-state batteries. Enhancing their performance necessitates simultaneous improvements in their stability and lithium conductivity. These properties can be calculated using first-principles simulations, provided that the crystal structure of the material and the diffusion pathway through the material are known. However, solid electrolytes typically incorporate dopants to enhance their properties, necessitating the optimization of the dopant configuration for the simulations. Yet, performing such calculations via the first-principles approach is so costly that existing approaches usually rely on predetermined dopant configurations informed by existing knowledge or are limited to systems doped with only a few atoms. The proposed method enables the optimization of the dopant configuration with the support of neural network potential (NNP). Our approach entails the use of molecular dynamics to analyze the diffusion after the optimization of the dopant configuration. The application of our approach to Li10MP2S12-xOx (M = Ge, Si, or Sn) reproduce the experimental results well. Furthermore, analysis of the lithium diffusion pathways suggests that the activation energy of diffusion undergoes a percolation transition. This study demonstrates the effectiveness of NNPs in the systematic exploration of solid electrolytes.
Read moreThe Role of External Donors in Ziegler−Natta Catalysts through Nudged Elastic Band Simulations on Realistic-Scale Models Employing a Universal Neural Network Potential
Masaki Fushimi and Devaiah DammaThis study undertakes a thorough computational exploration of Ziegler–Natta catalysis, emphasizing the role of external donors, particularly dicyclopentyldimethoxysilane (D donor), in the production of polypropylene. Employing the PreFerred Potential (PFP) model within the Nudged Elastic Band (NEB) method and Universal Neural Network Potentials (UNNP), we meticulously assessed the structural integrity of MgCl2 crystals, the dynamics of TiCl4 adsorption, and the kinetics of propylene insertion reactions. Our results demonstrated the precision of the PFP model in accurately replicating the crystalline structures and reaction mechanisms inherent in Ziegler–Natta catalyst systems.A pivotal finding from our research is the significant reduction in activation energy for isotactic propylene insertion, attributed to the presence of at least two D donors around a single Ti active site. Additionally, our computational approach, characterized by its speed and efficiency, successfully incorporates realistic catalyst models, encompassing a range of donor compounds, thereby bridging the gap between theoretical predictions and experimental practices. Our study not only corroborated the existing computational models but also provided novel insights into the mechanistic roles of external donors in Ziegler–Natta catalysis. The implications of these findings extend beyond theoretical studies, offering practical applications in the field of catalytic science and propylene polymerization. This research paves the way for future investigations, potentially transforming our understanding and utilization of Ziegler–Natta catalysts in industrial applications.
Read moreA New Zinc Salt Chemistry for Aqueous Zinc-Metal Batteries
Haoran Du, Yanhao Dong, Qing-Jie Li, Ruirui Zhao, Xiaoqun Qi, Wang-Hay Kan, Liumin Suo, Long Qie, Ju Li, Yunhui HuangAqueous zinc-ion batteries (ZIBs) are promising energy storage solutions with low cost and superior safety, but they suffer from chemical and electrochemical degradations closely related to the electrolyte. Here, a new zinc salt design and a drop-in solution for long cycle-life aqueous ZIBs are reported. The salt Zn(BBI)2 with a rationally designed anion group, N-(benzenesulfonyl)benzenesulfonamide (BBI−), has a special amphiphilic molecular structure, which combines the benefits of hydrophilic and hydrophobic groups to properly tune the solubility and interfacial condition. This new zinc salt does not contain fluorine and is synthesized via a high-yield and low-cost method. It is shown that 1 m Zn(BBI)2 aqueous electrolyte with a widened cathodic stability window effectively stabilizes Zn metal/H2O interface, mitigates chemical and electrochemical degradations, and enables both symmetric and full cells using a zinc-metal electrode.
Read moreSpectroscopic and theoretical analyses of the reaction of SrO in molten chloride and fluoride salts
Dokyu Kang, Choah Kwon, Wonseok Yang, Seokjoo Yoon, Yunu Lee, James T.M. Amphlett, Sang-Eun Bae, Sangtae Kim, Sungyeol ChoiIn this paper, the reaction of SrO in molten chloride and fluoride systems is investigated using Raman spectroscopy, combined with density functional theory calculations. The formation of Sr4OCl6 was observed in various salt compositions, including LiCl, LiCl-KCl, and LiCl-LiF, at temperatures ranging from 298 K to 923 K. Thermodynamic calculations estimated that Sr4OCl6 is more stable than SrO and SrCl2, while no stable strontium oxyfluoride compound was found. The results were also supported by X-ray analyses: Extended X-ray absorption fine structure analysis indicated that the local structure of Sr in LiCl-KCl and LiCl-LiF consists of one oxygen and six chlorides, corresponding to a complex with a similar local structure of Sr4OCl6. Additionally, Sr4OCl6 was found in LiCl-LiF, and no strontium compound was observed in LiF-KF using X-ray diffraction. These results provide a comprehensive understanding of the dissolution structures of strontium oxyhalide in molten chloride and fluoride systems.
Read moreProton Conduction over the Zeolite with Surface Water Cluster for the Water Electrolysis at Neutral Condition
Keigo Tashiro, Taisei Saito, Kojiro Goto, Junki Masuda, Takumi Miyakage, Shuhei Shimoda, Takashi Toyao, Nao Tsunoji, Ken-ichi Shimizu, Hiroshige Matsumoto, Shigeo SatokawaElectrolytic hydrogen production from water at neutral pH was achieved by using novel proton (H+) conduction system assisted by zeolite. Experimental and computational insights demonstrated that defective silanol nest generated by dealumination played an important role in the formation of water clusters and the H+ conduction over the zeolites proceeded via exchange of H-bonding between water molecules in the cluster. In addition, a proper balance between hydrophilicity and hydrophobicity is required for effective H+ conduction and silanol nest afford the balance. Furthermore, the contribution of hydrophilicity of the zeolite for the adsorption of water molecules became more drastic at high temperatures. Water electrolysis efficiency was strongly dependent on the H+ conductivity over the beta-type zeolite. The electrolytic cell containing the zeolite has the potential to be applied to the new hydrogen production system.
Read moreExploration of the mechanical properties of carbon-incorporated amorphous silica using a universal neural network potential
Hiroki Sakakima, Keigo Ogawa, Sakurako Miyazaki, Satoshi IzumiC-incorporated amorphous silica (a-SiOC) is expected to be a significant dielectric film for miniaturized semiconductor devices. However, information on the relationship among its composition, atomic structures, and material properties remains insufficient. This study investigated the dependence of the elastic modulus on the C content in a-SiOC, employing a universal neural network interatomic potential to realize a high-accuracy and high-speed simulation of multicomponent systems. The relationship between elastic modulus and atomic network structures was explored by fabricating 480 amorphous structures through the melt-quenching method without predetermined structure assumptions. The bulk modulus increased from 45 to 60 GPa by incorporating 10% C atoms under O-poor conditions and 20% C atoms under O-rich conditions, respectively. This result is attributed to the formation of denser crosslinking atomic network structures. In particular, the C atoms bonded with the Si atoms with higher coordination under O-poor conditions, whereas they tend to bond with O atoms under O-rich conditions, breaking the SiO2 network. Large C clusters precipitated as the C fraction was increased under O-rich conditions. Gas molecules, such as CO and CO2, were also generated. These results are consistent with reported ab initio calculation results of the formation energies of C defects and gas molecules in SiO2. The findings suggest that realizing O-poor conditions during deposition is crucial for fabricating stronger dielectric films. Therefore, this work contributes to understanding the fabrication of stronger dielectric films and elucidating the underlying mechanism of C cluster formation.
Read moreExploration of elastic moduli of molecular crystals via database screening by pretrained neural network potential
Takuya TaniguchiThe elastic properties of molecular crystals are important for pharmaceutical and material applications, but calculating the elastic constant tensor through theoretical computations is computationally expensive. This study evaluated the feasibility of using neural network potentials, which are noted for their lower computational cost compared to theoretical calculations, in predicting the elastic moduli of molecular crystals. The calculated elastic moduli were sufficiently consistent with experimental values, outperforming Hartree–Fock calculations. It was found that this method is precise enough for predicting the Young's modulus in specific directions via nanoindentation measurements, and relationships between the magnitude of the Young's modulus and crystal structures were also discovered. Furthermore, the database screening of elastic moduli of molecular crystals using a pretrained neural network potential suggested crystals with large and small moduli.
Read moreBoron coordination and three-membered ring formation in sodium borate glasses: a machine-learning molecular dynamics study
Takeyuki Kato, Federica Lodesani, Shingo UrataClassical molecular dynamics (CMD) simulations that employ analytical force fields have been commonly utilized to investigate mechanical, chemical, and thermal properties of oxide glasses owing to their superior computational efficiency. Conversely, simple functional forms limit the accuracy in modeling complicated glass structures, specifically, in alkaline borate glasses, which exhibit boron coordination numbers that vary nonlinearly with changes in glass composition and temperature. Machine-learning potentials (MLPs), which are trained using datasets on energy and force evaluated via the density functional theory (DFT), are garnering significant attention as a novel simulation technology for enhancing the accuracy in modeling materials. Therefore, this study applied a universal MLP, PreFerred Potential (PFP) (trade-name: Matlantis), to model sodium borate glasses, and its accuracy was verified in reproducing boron coordination and ring structures by comparing its results to available experimental data. We found that PFP can quantitatively reproduce the boron coordination change with glass composition without any empirical correction, while the boron coordination in the melts at high temperatures is overestimated, even though the qualitative variation was better estimated than CMD simulations. Furthermore, the MLP could generate many 3-rings, unlike the analytical force-field. Accordingly, we demonstrated superior accuracy of the MLP in modeling alkaline borate glasses, while discussing the challenges faced in reproducing the elaborated microstructures in borate glasses.
Read moreEvolutionary search for superconducting phases in the lanthanum-nitrogen-hydrogen system with universal neural network potential
Takahiro Ishikawa, Yuta Tanaka, Shinji TsuneyukiRecently, Grockowiak et al. reported "hot superconductivity" in ternary or multinary compounds based on lanthanum hydride [A. D. Grockowiak et al., Front. Electron. Mater. 2, 837651 (2022)]. In this paper, we explored thermodynamically stable phases and superconducting phases in the lanthanum-nitrogen-hydrogen system (LaxNyH1−x−y, 0≤x≤1, 0≤y≤1) at pressure of 20GP. We rapidly and accurately constructed the formation-enthalpy convex hull using an evolutionary construction scheme based on density functional theory calculations, extracting the candidates for stable and moderately metastable compounds by the universal neural network potential calculations. The convex hull diagram shows that more than fifty compounds emerge as stable and moderately metastable phases in the region of ΔH≤4.4mRy/atom. In particular, the compounds are concentrated on the line of x=0.5 connecting between LaH and LaN. We found that the superconductivity is gradually enhanced due to N doping for LaH and the superconducting critical temperature Tc reaches 8.77K in La2NH with y=0.25. In addition, we predicted that metastable La2NH2 shows the highest Tc value, 14.41K, of all the ternary compounds predicted in this study. These results suggest that it is difficult to obtain the hot superconductivity in the La-H compounds with N at 20GPa.
Read moreIncreasing the Sodium Metal Electrode Compatibility with the Na3PS4 Solid-State Electrolyte through Heteroatom Substitution
Lieven Bekaert, Suzuno Akatsuka, Naoto Tanibata, Frank De Proft, Annick Hubin, Mesfin Haile Mamme, Masanobu NakayamaRechargeable batteries are essential to the global shift towards renewable energy sources and their storage. At present, improvements in their safety and sustainability are of great importance as part of global sustainable development goals. A major contender in this shift are rechargeable solid-state sodium batteries, as a low-cost, safe, and sustainable alternative to conventional lithium-ion batteries. Recently, solid-state electrolytes with a high ionic conductivity and low flammability have been developed. However, these still face challenges with the highly reactive sodium metal electrode. The study of these electrolyte-electrode interfaces is challenging from a computational and experimental point of view, but recent advances in molecular dynamics neural-network potentials are finally enabling access to these environments compared to more computationally expensive conventional ab-initio techniques. In this study, heteroatom-substituted Na3PS3X1 analogues, where X is sulfur, oxygen, selenium, tellurium, nitrogen, chlorine, and fluorine, are investigated using total-trajectory analysis and neural-network molecular dynamics. It was found that inductive electron-withdrawing and electron-donating effects, alongside differences in heteroatom atomic radius, electronegativity, and valency, influenced the electrolyte reactivity. The Na3PS3O1 oxygen analogue was found to have superior chemical stability against the sodium metal electrode, paving the way towards high-performance, long lifetime and reliable rechargeable solid-state sodium batteries.
Read moreCO Adsorption on Ternary Nanoalloys by Universal Neural Network Potential
Ayako TAMURA, Gerardo VALADEZ HUERTA, Yusuke NANBA, Kaoru HISAMA, Michihisa KOYAMAMulti-element alloy nanoparticles have attracted attention for their potentially high catalytic properties. However, a high degree of freedom in configurations of metal atoms within nanoparticle increases the distinct adsorption sites, making it difficult to theoretically analyze its catalytic properties because the first-principles calculation requires a considerable computational cost. In this study, we develop a sequential scheme to calculate hundreds of adsorption sites by employing a pre-trained universal neural network potential named PFP. Our automated scheme is applied to CO single-molecule adsorption of CO onto PtRuIr ternary alloy nanoparticles. The calculation results are first compared with DFT results to confirm the accuracy. Adsorption energies in the alloy systems are widely distributed in comparison with those of the monometal counterparts, indicating that the alloy nanoparticle includes adsorption sites with various catalytic activities.
Read moreUsing GPT-4 in Parameter Selection of Materials Informatics: Improving Predictive Accuracy Amidst Data Scarcity and ‘Ugly Duckling’ Dilemma
Kan Hatakeyama-Sato, Seigo Watanabe, Naoki Yamane, Yasuhiko Igarashi, Kenichi OyaizuMaterials informatics and cheminformatics struggle with data scarcity, hindering the extraction of significant relationships between structures and properties. The "Ugly Duckling" theorem, suggesting the difficulty of data processing without assumptions or prior knowledge, exacerbates this problem. Current methodologies don't entirely bypass this theorem and may lead to decreased accuracy with unfamiliar data. We propose using Open AI GPT-4 language model for explanatory variable selection, leveraging its extensive knowledge and logical reasoning capabilities to embed domain knowledge in tasks predicting structure-property correlations, such as the refractive index of polymers. This can partially overcome challenges posed by the "Ugly Duckling" theorem and limited data availability.
Read moreOn the Thermodynamic Stability of Alloys: Combination of Neural Network Potential and Wang-Landau Sampling
Tien Quang Nguyen, Yusuke Nanba, and Michihisa KoyamaThermodynamic properties and atomic configuration of PdRu alloy were investigated using Wang-Landau Monte Carlo method in combination with a newly-developed universal neural network potential. By using this new potential, excess energy of PdRu alloy was calculated. It is found that PdRu alloy in FCC lattice is unstable in the full range of alloy composition. This agrees with previous study based on density functional theory. The combined method was able to determine the configurational density of states, from which thermodynamic properties of the alloy were derived. It is found that when temperature increases, the excess free energy of the alloy is reduced, increasing the possibility of alloy mixing. Depending on the composition, transition peaks appear at finite temperatures where there are changes of preferable atomic arrangement due to the effect of temperature via the configurational entropy. In addition, the analyses on short-range order parameter and bond fraction show that PdRu alloy prefers to be in segregated form, where Pd and Ru are immiscible at low temperature, consistent with the experimental observations. The random mixing of Pd and Ru atoms in the form of solid-solution can occur at high temperature.
Read moreAssessing the Reactivity of the Na3PS4 Solid-State Electrolyte with the Sodium Metal Negative Electrode Using Total Trajectory Analysis with Neural-Network Potential Molecular Dynamics
Lieven Bekaert, Suzuno Akatsuka, Naoto Tanibata, Frank De Proft, Annick Hubin, Mesfin Haile Mamme, and Masanobu NakayamaRechargeable batteries play a central role in the global shift from fossil fuels to renewable energy. Since the commercial introduction of lithium-ion batteries in the early 1990s, recent progress is focused on the development of solid-state materials and new battery chemistries. Specifically, solid-state sodium ion batteries are an attractive alternative alongside lithium-based rechargeable batteries, with improvements in safety, lifespan, sustainability, and price. Critical to the battery performance are electrode–electrolyte interfaces since undesirable side-reactions often proceed between electrode and electrolyte materials. In addition, atomistic or electronic-level knowledge on the reactions at the interface is limited due to technical difficulties of experimental observation. Computational studies on interfacial reactivity, such as first-principles techniques, have also long been limited by computational limitations. Recent advances using neural-network potential molecular dynamics simulations are allowing significantly larger systems and longer timescales to be simulated at a significantly reduced computational cost without loss of accuracy. In this study, the chemical stability of the glass–ceramic Na3PS4 solid-state electrolyte with the sodium metal electrode is investigated through a combined total trajectory analysis computational and experimental approach. PS4 groups in the Na3PS4 material were found to decompose sequentially into PS3, PS2, PS, and phosphide and sulfide species through the insertion of sodium atoms. Whereas the decomposition is thermodynamically favored, it is kinetically hindered due to steric effects in the PS3 intermediate. Machine learning-assisted analysis was found to be able to visualize the reactivity tendencies of individual element types. The formed SEI layer exhibited a good chemical stability and a low electronic conductivity. These findings provide new design principles to optimize and develop new solid-state electrolytes with an increased chemical stability toward the sodium metal electrode.
Read moreQuantum Annealing Boosts Prediction of Multimolecular Adsorption on Solid Surfaces Avoiding Combinatorial Explosion
Hiroshi Sampei, Koki Saegusa, Kenshin Chishima, Takuma Higo, Shu Tanaka, Yoshihiro Yayama, Makoto Nakamura, Koichi Kimura, and Yasushi SekineQuantum annealing has been used to predict molecular adsorption on solid surfaces. Evaluation of adsorption, which takes place in all solid surface reactions, is a crucially important subject for study in various fields. However, predicting the most stable coordination by theoretical calculations is challenging for multimolecular adsorption because there are numerous candidates. This report presents a novel method for quick adsorption coordination searches using the quantum annealing principle without combinatorial explosion. This method exhibited much faster search and more stable molecular arrangement findings than conventional methods did, particularly in a high coverage region. We were able to complete a configurational prediction of the adsorption of 16 molecules in 2286 s (including 2154 s for preparation, only required once), whereas previously it has taken 38 601 s. This approach accelerates the tuning of adsorption behavior, especially in composite materials and large-scale modeling, which possess more combinations of molecular configurations.
Read moreEffect of HFO Refrigerants on Lubrication Characteristics (Part 2) -Adsorption Characteristics of Various Refrigerants on Nascent Iron Surfaces and Molecular Simulation Analysis-
Yuji Shitara, Tasuku Onodera, Shigeyuki MoriFollowing the previous paper focusing on tribological properties of HFO (hydro fluoro olefin) refrigerant, the adsorption behavior of refrigerants on the nascent iron surface was investigated experimentally and the adsorption structure, adsorption energy and dynamic process of chemical reaction of refrigerants were analyzed by a molecular simulation. The adsorption behavior on the nascent iron surface was highly dependent on the molecular structure of the refrigerant. HFO refrigerants with an unsaturated bond exhibited high adsorption activity, and halogen species also affected the adsorption activity. HFO showed higher adsorption activity than organic ester, phosphate ester and alkyl sulfide as model compounds of refrigerator oil. In adsorption simulation by neural network potential (NNP), HFO molecules showed large negative adsorption energy. To understand the mechanism for this stronger adsorption of HFO species, density functional theory calculation was conducted, and it showed that HFO adsorbs on iron surface by electron donation from the molecule and back-donation from iron surface. There was also a good correlation between the experimental adsorption activity and the NNP-obtained adsorption energy. MD simulation of molecule adsorbed on the nascent surface at temperature of 298 K was subsequently done using NNP technique. The results showed that CF₃CF = CH₂ (R1234yf) exhibits a distinct decomposition reaction with releasing F atoms and it generates several Fe-F bonds, meaning that precursor of iron fluorides forms on the surface. It is worthy to mention that a formation of the iron fluoride has been experimentally detected on friction track by using XPS in the first report of this study. It was concluded that the adsorption and tribochemical formation of iron fluoride from HFO are supported by molecular simulation performed in this study.
Read moreEffect of HFO Refrigerants on Lubrication Characteristics (Part 1) -Tribological Characteristics under Refrigerant Atmosphere and Adsorption Characteristics on Nascent Metal Surface-
Yuji Shitara and Masayuki MoriFocusing on HFO (Hydro Fluoro Olefin) refrigerant, which is expected to be applied as a green refrigerant, the effect of R1234yf on the tribological characteristics was compared with HFC (Hydro Fluoro Carbon) refrigerant R32. Tribological tests were carried out with and without a model lubricant containing triethyl phosphate as an EP additive. R1234yf showed a lower friction coefficient and better wear resistance than R32. XPS analysis of lubricated tracks revealed that metal fluoride was formed from R1234yf and R32 and higher amount of metal fluoride was observed on the track formed under R1234yf than that under R32. The frictional characteristics of R1234yf were affected by refrigerant pressure and temperature of specimen. The higher the pressure of refrigerant and the temperature of the specimen, the better the tribological characteristics were observed. In order to clarify adsorption of refrigerant on metal surface, scratching tests were carried out under refrigerant atmosphere using a mass spectrometer. Although R32 did not adsorb, R1234yf adsorbed on nascent steel surface formed by scratching. The adsorption rate of R1234yf increased linearly with scratching speed. Since propylene also adsorbed on nascent steel surface, π-electrons in R1234yf play an important role on the adsorption. It can be concluded that R1234yf adsorbs easily on nascent steel surface resulting in formation of iron fluoride which acts as a boundary lubrication layer.
Read moreInnovation in Molecular Simulation Technologies for Tribology Using Artificial Intelligence
Tasuku OnoderaIn this article, a newly developed universal neural network potential (NNP), enabling accelerated molecular simulations, and its application to tribological phenomena were reported. The NNP is based on a graph neural network theory and an original dataset including a huge number of the first-principles calculation results. Two application examples using this method were introduced. One was exploring geometry for a fundamental molecular adsorption of long-chain fatty acid, which is difficult to deal with the conventional first-principles calculations. This new technique successfully observed that coverage of fatty acids on metal surface largely influence on the adsorption structures, i.e., orientation angle and intermolecular interaction of long-chain alkyl group. The second application was a molecular dynamics simulation for understanding influence of base oil structures on adsorption behavior of an oiliness additive. The adsorption time of an oiliness additive in three types of base oil (normal paraffin, isoparaffin, and naphthene) was measured. It was found that an oiliness additive in isoparaffin phase showed the earliest adsorption time among the tested base oils. This study unveiled the adsorption mechanisms of the oiliness additive in the base oils: an oiliness additive can smoothly pass inside the isoparaffin-derived oil film with sparse structure and easily reaches metal surface. We concluded that a rapid tribo-material discovery, as well as analysis on molecular mechanism of tribological phenomena, is expected to be achieved by adapting universal NNP to molecular simulations.
Read moreComparison of Matlantis and VASP bulk formation and surface energies in metal hydrides, carbides, nitrides, oxides, and sulfides
Shinya Mine, Takashi Toyao, Ken-ichi Shimizu, and Yoyo HinumaGeneric neural network potentials without forcing users to train potentials could result in significantly acceleration of total energy calculations. Takamoto et al. [Nat. Commun. (2022), 13, 2991] developed such a deep neural network potential (NNP) and made it available in their Matlantis package. We compared the Matlantis bulk formation, surface, and surface O vacancy formation energies of metal hydrides, carbides, nitrides, oxides, and sulfides with our previously calculated VASP values obtained from first-principles with the PBEsol(+U) functional. Matlantis bulk formation energies were consistently ~0.1 eV/atom larger and the surface energies were typically ~10 meV/Å2 smaller than the VASP counterpart. Surface O vacancy formation energies were generally underestimated within ~0.8 eV. These results suggest that Matlantis energies could serve as a relatively good descriptor of the VASP bulk formation and surface energies.
Read moreMolecular dynamics of electric-field driven ionic systems using a universal neural-network potential
Kaoru Hisama, Gerardo Valadez Huerta, Michihisa KoyamaIonic transport under an electric field bias is the fundamental component of electrochemical devices and processes. A universal neural network potential with Bader charge prediction is integrated into a Langevin thermostat NVT simulation to realize a direct simulation to examine those ion dynamics under an external electric field that is scalable and adaptable for various systems. We calculated the ion conductivity of O2− ions in yttria-stabilized zirconia (YSZ) and protons in hydrochloric acid water solution. The conductivity of YSZ shows a tendency consistent with the one using a Buckingham potential tuned for the system. For HClaq, the proton hopping contributes to the higher conductivity of protons than the counter anion Cl−, suggesting that our method is a promising tool for the ionic system, including chemical reactions and either for solid or liquid.
Read moreCyber Catalysis: N2 Dissociation over Ruthenium Catalyst with Strong Metal-Support Interaction
Gerardo Valadez Huerta, Kaoru Hisama, Katsutoshi Sato, Katsutoshi Nagaoka, Michihisa KoyamaCatalysis informatics is constantly developing, and significant advances in data mining, molecular simulation, and automation for computational design and highthroughput experimentation have been achieved. However, efforts to reveal the mechanisms of complex supported nanoparticle catalysts in cyberspace have proven to be unsuccessful thus far. This study fills this gap by exploring N2 dissociation on a supported Ru nanoparticle as an example using a universal neural network potential. We calculated 200 catalyst configurations consideringthe reduction of the support and strong metal-support interaction (SMSI), eventually performing 15,600 calculations for various N2 adsorption states. After successfully validating our results with experimental IR spectral data, we clarified key N2 dissociation pathways behind the high activity of the SMSI surface and disclosed the maximum activity of catalysts reduced at 650 °C. Our method is well applicable to other complex systems, and we believe it represents a key first step toward the digital transformation of investigations on heterogeneous catalysis.
Read moreCalculations of Real-System Nanoparticles Using Universal Neural Network Potential PFP
Gerardo Valadez Huerta, Yusuke Nanba, Iori Kurata, Kosuke Nakago, So Takamoto, Chikashi Shinagawa, Michihisa KoyamaIt is essential to explore the stability and activity of real-system nanoparticles theoretically. While applications of theoretical methods for this purpose can be found in literature, the expensive computational costs of conventional theoretical methods hinder their massive applications to practical materials design. With the recent development of neural network algorithms along with the advancement of computer systems, neural network potentials have emerged as a promising candidate for the description of a wide range of materials, including metals and molecules, with a reasonable computational time. In this study, we successfully validate a universal neural network potential, PFP, for the description of monometallic Ru nanoparticles, PdRuCu ternary alloy nanoparticles, and the NO adsorption on Rh nanoparticles against first-principles calculations. We further conduct molecular dynamics simulations on the NO-Rh system and challenge the PFP to describe a large, supported Pt nanoparticle system.
Read moreFollow us





Moves to the selected area